
Abstract
Protein-based virtual screening is integral to the modern drug discovery process. Most protein-based virtual screening experiments are performed using docking programs. The accuracy of a docking program strongly relies on the incorporated scoring function used, which is based on various energy terms. The existing scoring functions deal with the energy terms that use the equal weight function or other weight functions, which do not depend on characteristics of the protein. To improve the existing methods, Lu and Wang proposed a protein-specific scoring function based on a regression analysis that was shown to have higher performance than the existing methods. In this study, we propose a protein-specific scoring approach to select potential ligands based on logistic regression analysis. The performance of our method was evaluated using the Directory of Useful Decoys docked data set, which contains 40 protein targets. The results showed that the proposed method can increase the enrichment factors for most of the 40 protein targets.
1. Introduction
In recent years, protein–ligand docking methodologies have been rapidly developed and play an important role in the design of new drugs. The main goals of these methods are binding affinity estimation, pose prediction, and aiding virtual screening (Jain and Nicholls, 2008). Moreover, when exploring large compound libraries, the method must be able to successfully verify binding from nonbinding proteins and to rank these ligands correctly in the database (Kolb and Irwin, 2009). At present, with advance in computational tools, the docking process can be performed using computer software, such as AutoDock (Morris et al., 1998), DOCK (Kuntz et al., 1982; Ewing et al., 2001), and GOLD (Verdonk et al., 2003), and besides, the other related programs have been summarized (Pagadala et al., 2017). In general, a useful docking process consists of two components: an efficient search algorithm and an appropriate scoring function. In this study, we focus on the scoring function of protein–ligand docking.
During the docking process, search algorithms are used to investigate numerous ligand conformations. Scoring functions are used to evaluate the quality of the docking poses and to guide the search methods toward relevant ligand conformations. A scoring function must be able to distinguish the observed binding modes and associate them with the lowest energy values of the energy landscape. The second goal of the scoring function is to classify ligands and decoys (or inactive ligands) properly. The most important goal of the scoring function is to predict the binding affinity and to rank compounds according to their estimated binding affinities.
Three main classes of scoring functions have been described in the related literature: force field-based, empirical-based, and knowledge-based functions (Wang et al., 2003; Huang et al., 2010). In general, force field-based functions are derived from a classical force field and consist of a sum of energy terms. Empirical-based functions determine the scoring function and estimate binding affinity by using a regression approach (de Azevedo and Dias, 2008). Knowledge-based scoring functions are developed based on the statistical analysis of interacting atom pairs from protein–ligand complexes with available three-dimensional structures (Velec et al., 2005; Muegge, 2006). Although many scoring functions have been studied, no universal scoring function exists with significant reliability and efficiency for all proteins. Several studies have suggested that the performance of scoring functions could be improved by changing scoring strategies (Feher, 2006; Houston and Walkinshaw, 2013). Another way to improve the accuracy of the binding affinity prediction is to use a rescoring approach for each target protein individually; for example, developing the protein-specific scoring function (Lu and Wang, 2012). The score of the protein-specific scoring function depends on the protein specifically, different proteins having different scores. Hence, this scoring function type is expected to be more efficient for determining all about the protein family (Lu and Wang, 2012).
In this study, we propose a procedure to determine a protein-specific scoring function based on logistic regression, which was developed based on some thorough studies (Lu and Wang, 2012). The Directory of Useful Decoys (DUD) data sets (Huang et al., 2006) was used to validate the proposed method.
2. Materials and Statistical Methods
2.1. The DUD data set
The DUD data set is a published data set providing active compounds and decoys for crystal structures of ligand–target complexes. The DUD is designed for evaluating docking programs. This data set contains 2950 active compounds for a total of 40 target proteins. In addition, for every ligand, the data set contains 36 decoys that have similar physicochemical properties, such as molecular weights, calculated logP, and the number of hydrogen bonding groups, but structurally dissimilar (Huang et al., 2006). The docking procedure was performed and validated on these 40 protein targets with all ligands and decoys by using the DOCK program (Meng et al., 1992).
2.2. Enrichment factor and existing statistical method
The docking enrichment factor (EF) can be applied to measure the potential of the docking calculations to determine true positives throughout the database. Especially, the EF is described with respect to a given percentage of the database screened. Let x be the percentage of the compounds screened:
where
where
In the DOCK program (Meng et al., 1992), four energy terms (electrostatic interaction energy, van der Waals interaction energy, polar component of the ligand desolvation energy, and apolar part of the cost of ligand desolvation energy) are typically used to build a scoring function. The four energy terms are denoted as
The compounds with a smaller value of Eq. (3) are chosen as potential ligands. Because of the different biological backgrounds for each protein, Lu and Wang (2012) suggested using a protein-specific scoring function by summing the four energy terms with unequal weights, demonstrated as follows:
where
2.3. Logistic regression analysis
In this study, we use the form:
as a protein-specific scoring function, where 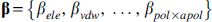 are unknown parameters. Consequentially, the compounds with smaller values of Eq. (5) are considered as potential ligands. For each protein, we proposed a method to estimate
are unknown parameters. Consequentially, the compounds with smaller values of Eq. (5) are considered as potential ligands. For each protein, we proposed a method to estimate  such that the scoring function [Eq. (5)] maximizes the EF value. We adopt the logistic regression method to estimate .
such that the scoring function [Eq. (5)] maximizes the EF value. We adopt the logistic regression method to estimate .
In logistic regression analysis, we first define the response variables Yi to be a decoy (
where  . We define a new scoring function by replacing with
. We define a new scoring function by replacing with 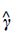 in Eq. (5):
in Eq. (5):
And then we select compounds corresponding to a small value of
Enrichment Factors for Each Protein Target Listed by the Equal Weight Scoring Function, Lu and Wang's Regression-Based Scoring Function, Logistic Regression Scoring Function, and Procedure 1 for the Top 1% Subset of the Entire Database
For each protein, we suppose that the ith smallest total energy of the n compounds for the protein is
where 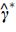 , which is an alternative estimator of , and we replace with in Eq. (7) to calculate the EF.
, which is an alternative estimator of , and we replace with in Eq. (7) to calculate the EF.
The reason that we use Eq. (8) to find an alternative estimator of is that we regard compounds with lower total energy based on the scoring function [Eq. (3)] to be an active compound. Thus, instead of using the true ligand directly, we denote that the response variable is zero corresponding to the compound with lower total energy based on the scoring function [Eq. (3)]. In other words, we treat the energy terms s and the corresponding EF value. The largest EF values and their corresponding estimations of are recorded. The details of this methodology for each protein are summarized in Procedure 1.
Procedure 1
Step 1
First, we assume that the compound with energy terms , and then define
for
Step 2
We assume that the compounds with energy terms is determined by using this logistic model [Eq. (8)], and the EF is then calculated as described in Step 1. We repeat the aforementioned step but change the values of response variables until there are s are calculated. The maximum of the EF and the corresponding are recorded. The corresponding is the desired coefficient. If more than one value is reached to the maximum value of EF, we randomly select one of them, and calculate .
3. Results and Discussion
In this study, the DUD data set is used as our data source and Procedure 1 is applied to obtain protein-specific scoring functions in virtual screening. A total of 40 proteins exist in the DUD data sets, and each protein has its own experimentally confirmed ligands. The number of ligands and decoys for each protein are listed in Table 1. To compare the performance of Lu and Wang's scoring function with the proposed method, we first evaluate the functions by using the top 1% compounds in the ranked database, as ranked by each scoring function. We compare the EFs of the equal weight scoring method [Eq. (3)], Lu and Wang's method [Eq. (4)], the logistic regression scoring method, and Procedure 1 for the top 1% compounds for each protein. The corresponding averages for the 40 proteins are 7.86, 16.49, 15.05, and 17.81, respectively. The logistic regression scoring method is slightly inferior to the Lu and Wang's method. However, Procedure 1 has a much better average EF than other methods.
In addition, we evaluate these methods for the top 20% of compounds in the ranked database (Table 2). The averages of the EFs for the top 20% compounds determined by using the equal weight scoring method, Lu and Wang's scoring method, logistic regression scoring method, and Procedure 1 are 2.21, 2.73, 3.35, and 3.36, respectively. These results demonstrate that both proposed methods provide more satisfactory results than the Lu and Wang's method.
Enrichment Factors for Each Protein Target Listed by the Equal Weight Scoring Function, Lu and Wang's Regression-Based Scoring Function, Logistic Regression Scoring Function, and Procedure 1 for the Top 20% Subset of the Entire Database
In Table 1, 32 of the 40 systems have EF values in Procedure 1 that are equally or much better than Lu and Wang's scoring method, although the improvement is not significant. In addition, as listed in Table 2, the logistic regression method and Procedure 1 show significant improvement compared with Lu and Wang's method. Although the logistic regression method provides inferior results compared with Lu and Wang's method for the top 1% compounds, it has the advantage of requiring less time. Therefore, the logistic regression method is a competitive method compared with Lu and Wang's method.
4. Conclusion
In this study, we adopt a method based on logistic regression analysis to increase the EF by developing a procedure for obtaining a protein-specific weight for energy terms. Our results show that this protein-specific scoring method could improve the equal weight scoring function and regression-based protein-specific scoring function for the 40 protein targets in the DUD data sets. It is also expandable to larger databases. Furthermore, this method is not limited to the DOCK scoring function. It can be applied to modify other scoring functions, such as the GOLD score and Glide score. We believe that this method can significantly elevate the hits rate, which can benefit the modern drug discovery process.
Footnotes
Author Disclosure Statement
The authors declare they have no competing financial interests.
Funding Information
This study was supported by the Ministry of Science and Technology, Grant No. 107-2118-M-009-002-MY2, Taiwan.