
Abstract
Accumulating evidence suggests that long noncoding RNAs (lncRNAs) are emerging as important regulators involved in diseases, including heart failure (HF). In this study, we used microarray profiles to examine the transcriptome of lncRNAs in left ventricle samples derived from HF patients. We designed a custom pipeline to reannotate lncRNAs from microarray data and identified a set of consistently dysregulated lncRNAs in HF across the three independent cohorts. In total, 84 lncRNAs were found to be consistently dysregulated in at least two cohorts. By using a rank aggregation method, we integrated correlated protein-coding genes of the consistently dysregulated lncRNAs derived from HF samples and characterized their biological functions based on the correlated genes. The transcriptional regulation relationships of these lncRNAs ranged from 104 to 261, suggesting their important regulatory functions. Among the conserved lncRNAs, AC018647.1 and AC009113.1 showed significant dysregulation across all three cohorts. Our results showed that the two lncRNAs were involved in development-associated and cardiac cycle-associated functions.
1. Introduction
Heart failure (HF) is a leading cause of morbidity and mortality worldwide (Henes and Rosenberger, 2016). With the exception of heart transplantation, there is no curative treatment strategy available. Gene expression reprogramming is thought to be the base of pathological cardiac hypertrophy and HF, reflecting the activation of heart development (Thum and Condorelli, 2015). Therefore, studies of the complex genetic regulation of maladaptive reprogramming in HF is essential for better understanding of HF pathogenesis, and could lead to new diagnostic and therapeutic tools.
For the past few years, high-throughput analysis of the transcriptome has discovered long noncoding RNAs (lncRNAs): these are a class of important regulatory molecules in gene expression and various diseases through various mechanisms (Batista and Chang, 2013; Yu et al., 2018a). Many studies have shown that the myocardial transcriptome is dynamically regulated in advanced HF and the expression levels of lncRNAs can discriminate failing hearts of different pathologies, showing lncRNAs are important regulators of cardiac gene expression and can significantly influence cardiac homeostasis and functions (Yang et al., 2014). For example, Kumarswamy et al. (2014) found that higher LIPCAR levels were associated with a higher risk of cardiovascular death, and showed this lncRNA to be an independent biomarker of cardiac remodeling and could predict the survival of patients with HF. Given the large number of lncRNAs detected, this represents a challenging research opportunity to understand their roles in HF.
In this study, by using transcriptome profiles offered by large independent HF cohorts, we detected >10,000 lncRNAs in HF and identified 84 consistently dysregulated lncRNAs in more than two cohorts. After integrating the coexpression relations between the dysregulated lncRNAs and the mRNAs, we further characterized the transcriptional regulation relationships of two lncRNAs that dysregulated in all cohorts and showed their roles in HF.
2. Methods
Heart failure data sets preparation
The transcriptome profiles from three HF studies were retrieved from the Gene Expression Omnibus (GEO), including 30 samples (15 HF samples and 15 matched controls) from GSE16499, 38 samples (30 HF samples and 8 controls) from GSE21610, and 8 samples (4 HF samples and 4 controls) from GSE21610. The Affymetrix Human Exon 1.0 ST Array and Affymetrix Human Genome U133 Plus 2.0 Array were used considering its relatively comprehensive coverage of lncRNAs.
Data processing
The lncRNA annotations were retrieved from GENCODE (v16). We designed a custom pipeline to reannotate lncRNAs according to previous studies (Du et al., 2013). The probe sequences were downloaded from the manufacturer's website (www.affymetrix.com) and uniquely mapped to the human reference genome (hg19) by Bowtie (Langmead et al., 2009). Then, BEDTools (http://code.google.com/p/bedtools) were used to identify probes completely falling into exons of lncRNAs and those did not overlap with protein-coding genes were retained to compute the expression levels of lncRNAs. Finally, lncRNAs that annotated by at least four probes were thought to be expressed in the sample. The raw intensities of the probes were normalized using the RMA normalization method and the expression alterations between HF samples and control were calculated by Student's t-test analysis.
Identification of protein-coding genes related to the long noncoding RNA
The association between the protein-coding genes and an lncRNA was assessed by the coexpression levels (Pearson correlation coefficients) calculated in HF samples in each of the three cohorts. The lists of protein-coding genes from the three cohorts were then ranked based on their correlation coefficients. To ensure that protein-coding genes can be ranked in a more reliable way (robustly related to the lncRNA), we used robust rank aggregation method (Kolde et al., 2012). This method assigns a p-value to each element in the aggregated list, which describes how much better it is ranked than expected. We applied the integrated approach to aggregate the individual ranks into one final rank based on order statistics (Pearson correlation coefficients). The regulatory relations between significantly relevant protein-coding genes and the lncRNA were displayed by Cytoscape 3.3.0 (Shannon et al., 2003).
Functional analyses
We performed functional enrichment analysis for the related protein-coding genes of the lncRNA signature using gene set enrichment analysis (GSEA). Functional gene sets were downloaded from MSigDB database and the final rank of related protein-coding genes was used as preranked gene list. p-Value of 1% was used as criteria for significantly enriched gene sets.
3. Results
Data description
To identify lncRNAs in abundant HF cohorts, we collected all microarray profiles in HF that were suitable to detect lncRNAs: (1) 15 HF samples with 15 age- and gender-matched control samples from GSE16499 (Kong et al., 2010); (2) 38 HF samples with 8 nonfailing control hearts from GSE21610 (Schwientek et al., 2010); and (3) 4 HF and nonfailing normal pairs from GSE76701 (Kim et al., 2016) (Table 1). All of the three cohorts were collected from left ventricle to ensure the comparison would not be affected by tissue sampling. Two types of microarrays (GPL570 and GPL5175) were used because of their substantial amount of probes to detect lncRNAs. After the probes from microarray were reannotated, we annotated 2673 and 10092 lncRNAs for GPL570 and GPL5175, respectively.
Data Sets of Heart Failure Used in the Study
Transcriptome analysis reveals consistently dysregulated long noncoding RNAs in heart failure
We identified 785, 457, and 139 differentially expressed lncRNAs between HF samples and control for these three cohorts, respectively (p < 0.05, see Materials and Methods section). Across three cohorts, 84 lncRNAs were found to be consistently altered in at least two cohorts (Fig. 1A and Supplementary Table S1) and 2 lncRNAs were consistently dysregulated in all three cohorts (i.e., AC018647.1 and AC009113.1, Fig. 1B). Some of them have been shown to have important roles in HF, such as DANCR (Zhang et al., 2015), XIST (El Azzouzi et al., 2016) and SNHG1 (Zhang et al., 2018). However, for most dysregulated lncRNAs, their functional characterizations were lacking.

Differentially expressed lncRNAs in the three HF cohorts.
Identification of the transcriptional regulation relationships of the consistently dysregulated long noncoding RNAs
Guilt-by-association is widely used to characterize lncRNAs by exploiting other biological contexts (Signal et al., 2016). By calculating the degree of coexpression between each protein-coding gene and the consistently dysregulated lncRNAs in HF, we identified the correlation of protein-coding genes to the lncRNAs from the three cohorts, respectively (one lncRNA [AC103706.1] was excluded because of its absence in GSE16499). To identify the transcriptional regulation relationships among lncRNAs and protein-coding genes, we further used the robust rank aggregation method (Kolde et al., 2012) to integrate the correlation results across the three data sets (Fig. 2A). The rank-based method is proper to solve the problem of the heterogeneity among different high-throughput genomic experiments. An average of 161 transcriptional regulation relationships were found for each lncRNA (ranged from 104 to 261, Fig. 2B). Significant scores showed that 170 statistically relevant genes were found to AC018647.1 and 149 statistically relevant genes were found to AC009113.1 (p < 0.01, Fig. 2C). We also found that two protein-coding genes (i.e., OR51E1 and RAB9B) were related to both lncRNAs. OR51E1, which was a member of G-protein-coupled receptor family, was proved as the most highly expressed odorant receptor in cardiac development (Jovancevic et al., 2017).
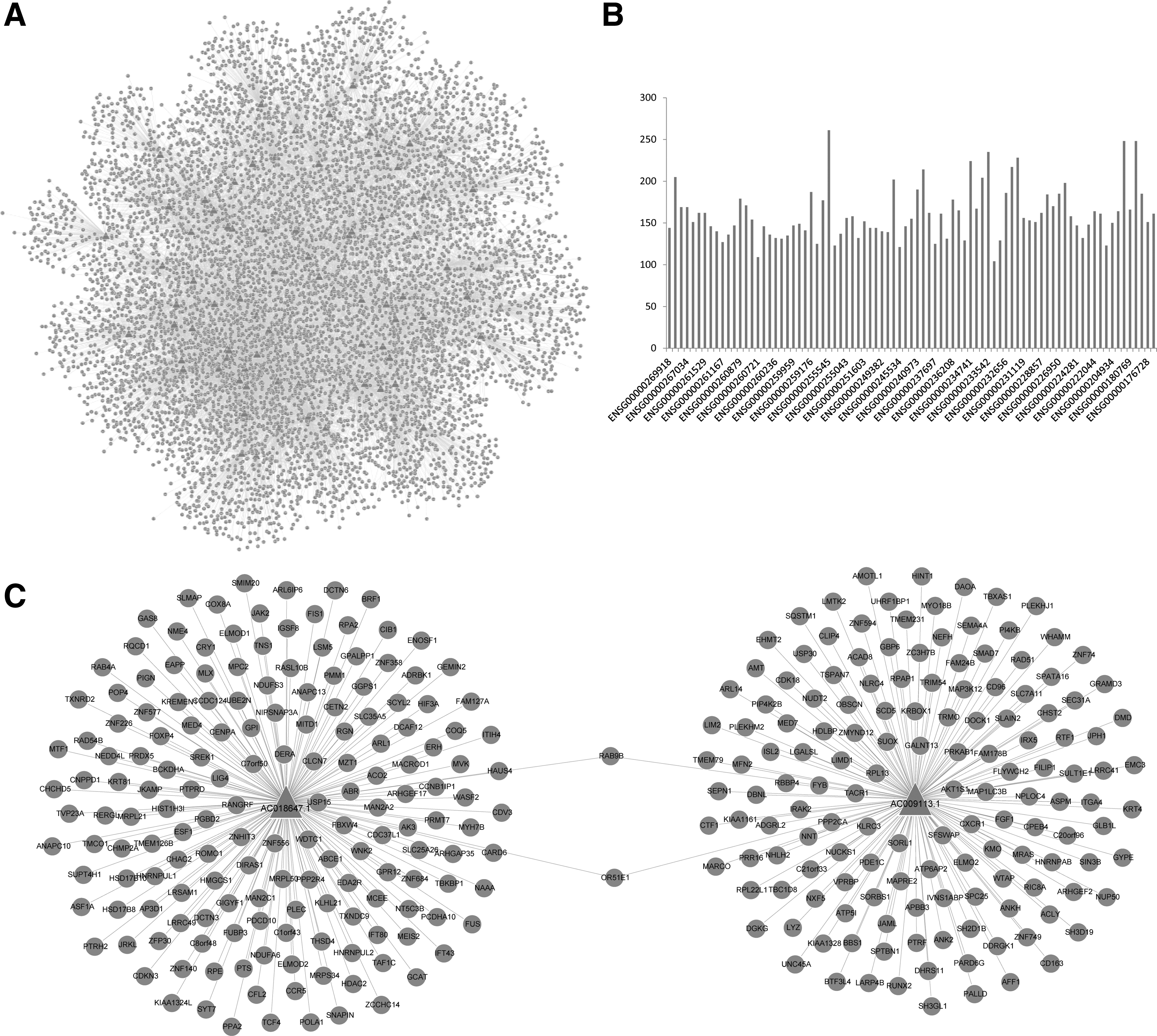
Transcriptional regulation relationships of the consistently dysregulated lncRNAs in heart failure.
The two dysregulated long noncoding RNAs involved in important functions in heart failure
Based on the transcriptional regulation relationships of protein-coding genes, we used the GSEA to identify biological processes associated with the consistently dysregulated lncRNAs on the basis of the significant score. Some known functions related to HF were identified, such as regulation of cardiac muscle contraction and ventricular cardiac muscle cell differentiation. Figure 3 displayed the functional results of the two dysregulated lncRNAs. AC018647.1 was significantly enriched in labyrinthine layer blood vessel development, negative regulation of insulin secretion and negative regulation of peptide hormone secretion (Fig. 3A), and AC009113.1 was significantly enriched in positive regulation of heart rate, positive regulation of heart contraction, and activation of protein kinase B activity (Fig. 3B).
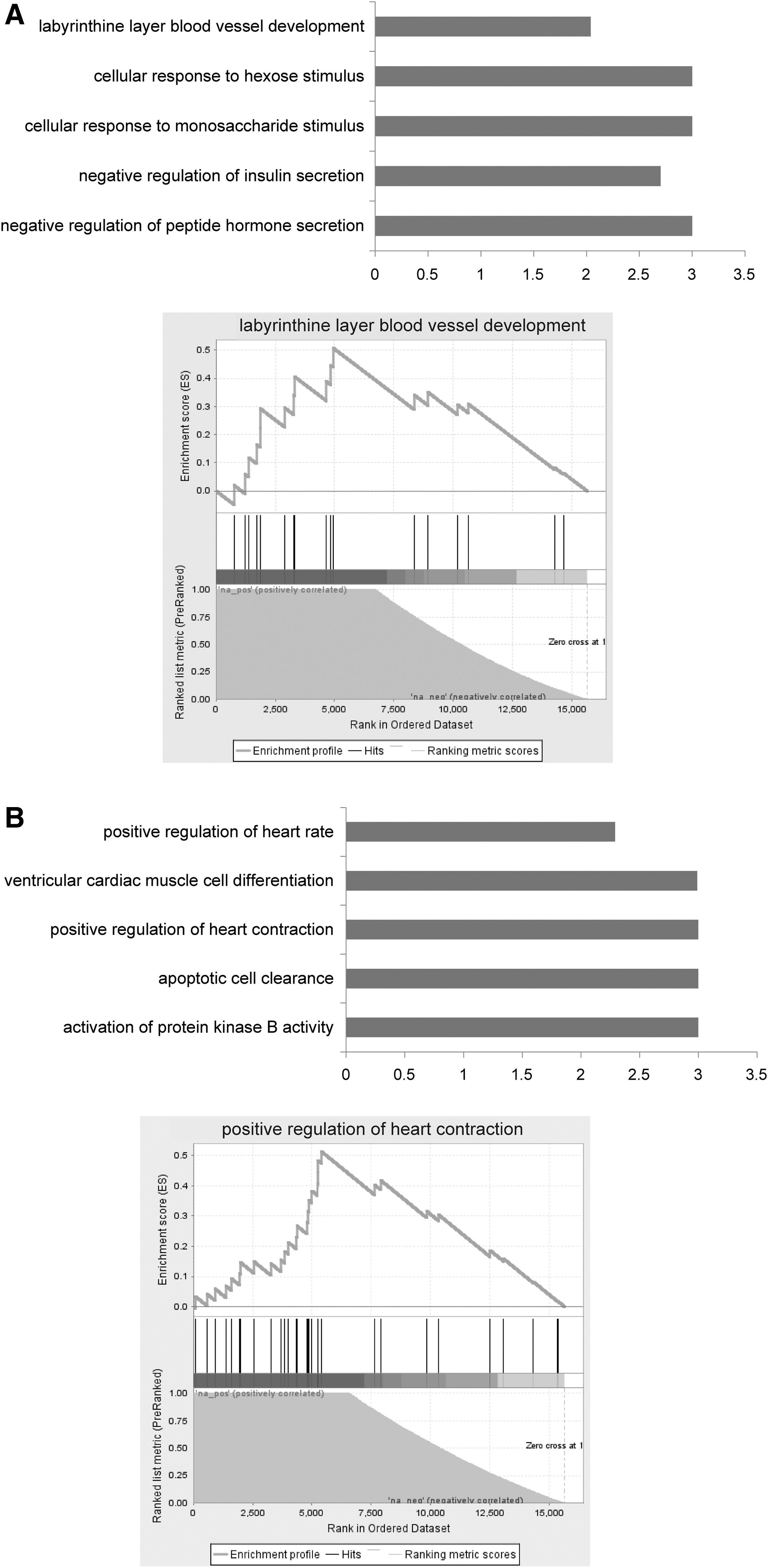
Functional analysis of the two consistently dysregulated lncRNAs. Gene ontology enrichment and enrichment plot of the correlated protein-coding genes of AC018647.1
4. Discussion
HF is one of the biggest contributors to human morbidity and mortality. During the past few years, the discovery of thousands of lncRNAs has provided new insights into the mechanisms involved in human HF. Functional modulation of ncRNAs in cardiovascular disease animal models has demonstrated their importance, which facilitated researchers to develop novel ncRNA-based therapeutic strategies (Dangwal and Thum, 2014; Yu et al., 2018b). In this study, we used an unbiased approach to comprehensively examine the lncRNA profiles in HF across three independent cohorts. We identified 84 HF-associated lncRNAs that consistently dysregulated in at least two cohorts and characterized the transcriptional regulation relationships based on their integrated correlations of protein-coding genes in HF samples. Our results showed that the two consistently dysregulated lncRNAs were involved in development-associated and cardiac cycle-associated functions, suggesting that the integrated transcriptome analysis could accurately reflect the physiological and pathological characteristics of the failing heart.
GSEA highlighted several important cardiovascular functions enriched in HF lncRNAs-correlated protein-coding genes (Lopes and Elliott, 2013; van Berlo et al., 2013). The analysis fits well with previous studies of functional characterization of HF-associated lncRNAs, suggesting that the dysregulated lncRNAs identified in this study might have an important role in HF progression and prompting validation with more direct approaches (Li et al., 2013; Greco et al., 2016).
A therapeutic strategy for HF is to cure the sick heart cells by interfering with the gene expression program that underlies HF. RNA-based strategies might become novel therapeutic strategies for HF by interfering with the gene expression program that underlies HF (Papait et al., 2013). The potential of this strategy for HF has been supported by the possibility of manipulating ncRNAs in vivo to trigger the gene expression program of HF (Care et al., 2007; van Rooij et al., 2008). Thus, lncRNAs could open new therapeutic opportunities for HF, which would also improve our understanding of the ncRNA network involved in regulating gene expression changes underlying HF. Our study provided a set of consistently dysregulated lncRNAs in HF, which could be regarded as potential therapeutic targets. Before these opportunities can become real, it is necessary to future investigate the identified HF lncRNA actions and their capacity to predict the pathology.
Our study presented consistently dysregulated lncRNAs in HF by employing large independent patient cohorts, suggesting a conserved lncRNA mechanism in HF progression. Our results facilitated understanding the molecular mechanisms underlying HF progression and suggested that lncRNAs may have the potential for HF diagnosis and treatment.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.