
Abstract
Next-generation sequencing technologies are producing genomic data at ever-increasing rates. It has become a challenge to store, transmit, and process the massive quantity of data, creating a vital need for a tool that compresses genomic data produced in a lossless manner, thus reducing storage space and speeding up data transmission. Data centers are adopting either of the two general-purpose genomic data compressors: gzip or bzip2. Both these use Huffman encoding, although they implement it in different ways. However, neither of these two takes advantage of properties of DNA data, such as the presence of a small alphabet and many repeats. Huffman encoding compression can be improved by exploiting DNA characteristics. Recently, it has been shown that Huffman encoding compression can be improved by creating an unbalanced Huffman tree (UHT), which demonstrates significant advances in compression over a standard Huffman tree used in both gzip and bzip2. However, the UHT created is greedy. This article proposes an improved nongreedy UHT (NUHT), a lossless nonreference-based fasta and multifasta compressor. We compare our algorithm with two well-known general-purpose compressors, gzip and bzip2, as well as with UHT, a DNA-specific compressor based on Huffman tree. Our algorithm outperforms all three in terms of compression ratio and is seven times faster than UHT.
1. Introduction
Currently, data deluge has become the main issue affecting most genomic centers. Owing to the development of high-throughput sequencing technologies, massive volumes of genomic data are being rapidly and inexpensively generated every day, creating two main challenges: data growth and bandwidth required to transmit these data (Muir et al., 2016). Compression tackles both challenges by reducing both issues: storage space required by genomic data and bandwidth used to transmit data over the network (Kahn, 2011; Giancarlo et al., 2013; Alberti et al., 2015).
Data compression is the art of reducing the bits required to represent data without detectable effects on the representation of the original form. Numerous algorithms have been developed for all types of data, including text, image, audio, and video. However, all of these algorithms are either lossy or lossless. Lossy compression may eliminate insignificant or redundant bits of data when compressing audio, video, and image data, and it is appropriate when the exclusion of some data does not have a crucial effect on representation of the content. Lossless algorithms, on the other hand, prevent data loss of any sort and guarantee full recovery of data after decompression. Lossless algorithms for text have been well studied, and recently, many such algorithms, including gzip, bzip2, Lempel–Ziv (LZ), and Lempel–Ziv–Welch (LZW), have been developed. Text compression using these algorithms works well for languages with large alphabets, such as English. However, these tools were not specifically developed to optimize text compression by utilizing biological sequence characteristics such as small alphabet size (i.e., the four-letter alphabet of DNA contains only nucleotides A [adenine], T [thymine], C [cytosine], and G [guanine]) or some meaningful biological features, such as repeats and palindromes (Giancarlo et al., 2012; Bakr and Sharawi, 2013; Zhu et al., 2013; Wandelt et al., 2014). Thus, there is scope for creation of more targeting algorithms.
Many studies on genomic data compression have taken these characteristics of sequences into account, leading to algorithms designed specifically to compress genomic data. Such special-purpose algorithms can be categorized as either reference-free or reference-based algorithms. Reference-free algorithms are based on characteristics of the targeted sequences, while reference-based algorithms need a set of reference sequences to be used in their method of compression. Such algorithms align the targeted sequence to its reference and then encode their mismatches. The decompressor then must have access to the reference sequence(s) used in the compression process. The reference-based methods usually obtain high compression ratios, but both the compressor and decompressor must have the same reference sequence(s).
The first reference-free algorithm developed was Biocompress (Grumbach and Tahi, 1993). This algorithm is based on the compression method of Ziv and Lempel (1977), in which repeats and palindromes in the sequence are found and then encoded using the length and position of their earliest occurrence. Biocompress-2 (Grumbach and Tahi, 1994) improves upon the compression of Biocompress by implementing arithmetic coding of order-2 in its method. Cfact (Rivals et al., 1996), another reference-free algorithm, comprises two phases: parsing and encoding. In the parsing phase, a suffix tree data structure is built to find the most significant repeats. In the encoding phase, the first occurrence of repeats and all nonrepeats are encoded using two bits per base. Later occurrences of repeats are encoded using pointers to their first instance. Approximate repeats are used for data compression and were first implemented in the GenCompress algorithm (Chen et al., 1999), which uses the position and length of noninitial occurrence of repeats in the encoding stage. Building upon GenCompress, the DNACompress algorithm (Chen et al., 2012) was designed to exploit data by first finding all approximate repeats containing palindromes using the PatternHunter search tool (Ma et al., 2002) and then encoding the approximate repeats and nonrepeats by implementing the Ziv and Lempel compression method (1977).
Based on the normalized maximum likelihood model, the NMLComp algorithm (Tabus et al., 2003) utilizes a modified version of discrete regression to better encode the approximate repeats by combining the regression with the first-order Markov model. The next algorithm specialized for DNA encoding was the GeNML algorithm (Korodi and Tabus, 2005), which is an improved version of NMLComp. The methodology used reduced the cost of searching for approximate repeat matches and the ability to choose the block size used in parsing the target sequence. The DNACompact algorithm (Gupta and Agarwal, 2011) uses a word-based tagged code (WBTC). It first converts the target sequence into words in such a way that the four-symbol alphabet is transformed into a three-symbol alphabet. The sequence is then encoded using WBTC. The DNACompact algorithm is based on pattern-aware contextual modeling, which first searches for exact repeats and palindromes and then represents them by a compact quadruplet. Then, it uses nonsequential contextual models to exploit the features of DNA sequences and uses logistic regression to synthesize these models.
Based on Huffman trees, the unbalanced Huffman tree (UHT) (Al-Okaily et al., 2017) was recently introduced: it utilizes the notion of skewed or UHTs. This algorithm comprises three phases: (1) collecting the k-mer; (2) ensuring that the Huffman tree is unbalanced; and (3) encoding. The next category of algorithms that we will discuss comprises algorithms that use a reference-based method. As the human genome is 99% self-similar, the DNAzip algorithm (Christley et al., 2008) takes advantage of this similarity and considers only the variation in the targeted sequence to compress data, and then uses the reference for decompression. The relative Lempel-Ziv (RLZ) algorithm (Kuruppu et al., 2010) uses the LZ77 approach to parse each genome into factors, using the reference sequence as a dictionary, for compression. The genome resequencing (GRS) algorithm (Wang and Zhang, 2011), based on the diff tool in UNIX, first looks at the reference and target sequences for the longest subsequence that is identical in both. Then, a similarity measure is calculated and compared with a threshold; if it is larger, data are compressed using Huffman encoding; otherwise, they are divided into smaller subsequences and the previous steps are applied again. GREn (Pinho et al., 2011) is a statistical compression-based method that uses a probabilistic copy model. In this algorithm, the model estimates the probability distribution of each symbol, considering that the target sequence is a copy of the reference sequence. The probability distributions are then sent to the encoder.
2. Approach
Huffman encoding has been a popular algorithm to compress data with variable-length codes. Since its development in 1952, the algorithm is used in well-known general-purpose compressors, such as gzip and bzip2. Since each character is 8-bit long, Huffman coding assigns variable-length codes to each character such that the lengths of the codes are based on the number of occurrences of the character in the text. In this case, the most frequent character will have the shortest code, while the least frequent character will have the longest code. For example, given a DNA sequence, ACAATGGTGACAGCACGGGCTAAAGCTTCGATGGAAAAAA, which has 40 characters, the binary representation of each base is as follows: A = 01000001, C = 01000011, G = 01000111, and T = 01010100. The memory space occupied by this DNA sequence is (40 × 8) = 320 bits.
We summarize the steps required to compress this sequence using Huffman encoding. The algorithm will count the frequency of each base and create a node that associates each base with its count. Then, these nodes will be used to create a binary tree. First, the two bases with the incidence counts will be merged in a parent node. The incidence count of the new parent node is the sum of the counts of the two children. Then, the process will be repeated, with the new parent node treated like the others. Again, the two nodes with the least incidence counts will be merged. This process will continue until no node is left. Finally, a binary tree will be built with left edges labeled 0 and right edges labeled 1 for each node. To find the new base representation, the binary tree will be traversed from the root down to the desired leaf (base). In Figure 1, the path from the root down to base C provides the new bit representation 111, while the path to reach base A from the root is 0. For the other bases, see Table 1.

Huffman tree.
Old and New Bit Representation
The compressed sequence represents each base using fewer bits than those for each base depending on their frequency in the original sequence. Instead of being represented by 8 bits, the new bit code lengths in our example are 1, 2, 3, and 3 for A, G, C, and T, respectively. To calculate the obtained compression ratio, the new bit count of the sequence is divided by the original count. In this case, the compression ratio = (1 × 16) + (2 × 11) + (3 × 6) + (3 × 7)/320 = 24%.
Recently, to yield a better compression ratio, a tool called UHT, which is built on the concept of Huffman encoding, was proposed. UHT considers DNA sequence characteristics when constructing the Huffman tree and forces the resulting tree to be unbalanced. The UHT algorithm surpasses the conventional Huffman tree in terms of compression and has been tested against gzip and bzip2. However, we found that UHT was created greedily and can thus be improved.
3. Methodology
Our algorithm works by creating a nongreedy UHT (NUHT) for DNA sequence compression. The regular Huffman tree and the UHT are created greedy and thus not able to choose the optimal k-mer and bases, which (by encoding the compressor) gain a better compression ratio. For this purpose, our algorithm first restricts the k-mer length to 2 because if the algorithm allows for another k-mer, such as 3-mer or 4-mer, then the 2-mer will need more than 3 bits for its encoding, and this, in general, will decrease the compression ratio gain since the 2-mer has a large effect on compression because it results in 2-mers (represented by 16 bits) being encoded by only 3 bits. After that the algorithm runs to find the frequency of all 2-mers and the frequency of each base (A, C, G, and T). Since the algorithm works by first encoding the 2-mer and then encoding the bases, this changes the counted base frequency, so our algorithm first finds the optimal 2-mer that (by encoding) leads to minimizing the lowest numbered base count, which has a high impact on the compression ratio because it encodes a single base that is represented by eight bits to three bits and thus has a bad reflection on the compression gain in general and thus needs to be minimized. After that our algorithm builds the NUHT, and the base with the least frequency count and the optimal 2-mer are encoded using only three bits, while the other three bases are encoded using two bits.
The NUHT can be created through the following steps:
First, the frequency of each base (A, C, G, and T) is calculated. At the same time, the non-ACGT bases and the starting positions of the N bases and their ending positions are marked. The base that appears least frequently is determined using Step 1. The frequency of each k-mer (in this case, k = 2) is collected. Based on the frequency of each 2-mer, each base (A, C, G, and T) in the collected 2-mers is weighed. If a single 2-mer consists entirely of the same base (such as AA, CC, and so on for 2-mer), then its weight will be multiplied by 2 by the 2-mer frequency. Finally, the base from each one with the highest weighted value is chosen. The weighted values of bases from those calculated in Step 1 are negated. Using the result from Step 5, the base with the next lowest value is found. A UHT based on the above using four bases and one 2-mer is built. The four bases and one 2-mer are encoded with their Huffman code generated in the previous step. The encoded sequence is converted into ASCII code with the addition to the headers, which include information about the sequence, such as the code word, positions of non-ACGT bases, and starting and ending positions of the N bases, for later use when decoding the sequences.
Figure 2 depicts the NUHT after the final step. The tree has three bases assigned to two bits with ratio impact of 25%, one base assigned to three bits with ratio impact of 37.5%, and one 2-mer assigned to three bits with ratio impact of 18.75%; see Table 2.
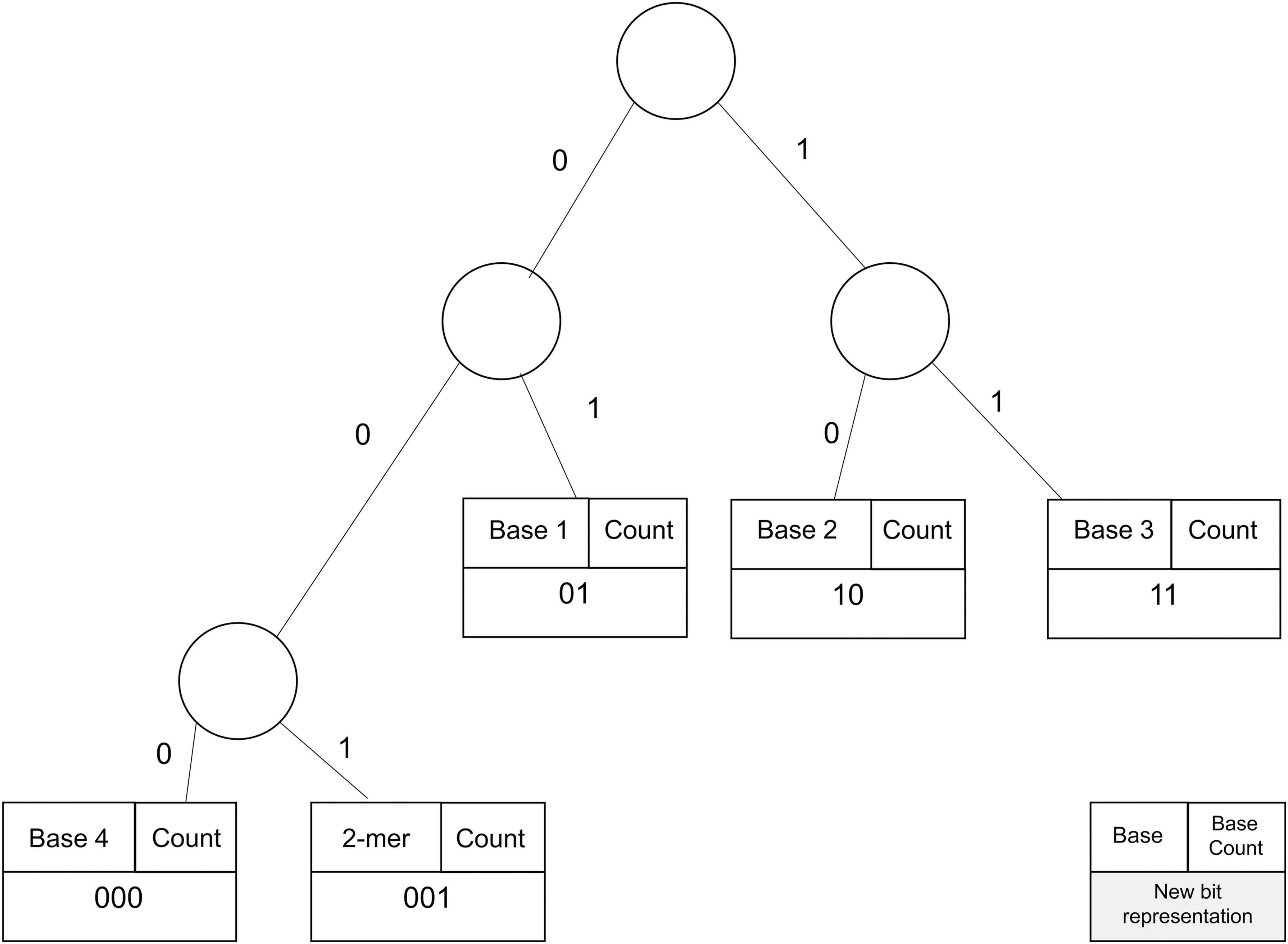
Nongreedy unbalanced Huffman tree.
Compression Ratio Impact of Bases
All NUHTs have the same shape and will be used to compress all different DNA sequences; the shape will not be affected by the targeted DNA sequence since it is used to create the optimal Huffman tree for such small alphabets. The main goal of previous steps of the nongreedy Huffman tree algorithm is to minimize the frequency count of the least base (base 4) and maximize that of the 2-mer. This leads to a better compression ratio based on the compression ratio impacts; the ratio impact is calculated by the following equation:

4. Datasets
We used the dataset used by Al-Okaily et al. (2017), which consists of five different species: Cholerae, Abscessus, Saccharomyces, Neurospora, and Chr22 (NCBI, 2001; 2008; 2011; 2013; 2015). The sequence data are in fasta and multifasta formats with extension.fasta. The fasta format provides text-based data, which include the DNA sequence information in the first line as a header, followed by lines of varying lengths (typically 70 bases). The multifasta format is similar to the fasta format, but it can contain more than a single sequence; in multifasta, there are multiple header lines across the genome.
5. Results
We implemented a nongreedy Huffman tree algorithm using Python with no parameters. Our tool was compared with other tools that utilize Huffman tree encoding, namely gzip, bzip2, and various implementations of UHT. The first two are well-known general-purpose tools used by data centers to compress genomic data, while the third one is a special-purpose compressor for DNA sequences. gzip can compress DNA sequences as well as various data types and run on different platforms. It is based on the LZ algorithm and utilizes Huffman encoding in its core. When compressing data, gzip builds two Huffman trees: the first one compresses the match lengths of a duplicated string and the second one is for the distance. The gzip compression ratio is generally around 30%. Another popular compressor, bzip2, stacks several layers of compression techniques, one of which is Huffman encoding; the compression ratio of bzip2 tends to be better than that of gzip, mostly reaching around 28%. Finally, there are three tools based on UHT: the basic UHT, a UHT that prioritizes encoding k-mers containing the least frequent base (UHTL), and multiple UHTL (MUHTL). As mentioned by Al-Okaily et al. (2017), all UHT implementations show improvement over the conventional Huffman trees used in both gzip and bzip2.
When comparing our algorithm with the tools described above, all experiments were performed on the same machine running 64-bit Ubuntu with a 2.6-GHz Intel Core i5 processor and 4-GB memory. Compared with gzip, bzip2, and the basic implementation of UHT, our tool delivered the best for all data used in Table 3. Our algorithm, NUHT, demonstrated significant improvement over conventional Huffman tree encoding and slight improvement over the basic UHT. We also compared our algorithm with improved implementation of UHT, namely UHTL and MUHTL, both of which show slight improvement over the basic UHT, but the greedy behavior of UHTL and MUHTL prevented the achievement of better compression ratios than those achieved by our algorithm (Table 4).
Compression Ratio Defined as the Ratio of the Compressed File Size to the Original File Size
Bold font indicates that a value has the best compression ratio among those tested for the given sequence.
NUHT, nongreedy unbalanced Huffman tree; UHT, unbalanced Huffman tree.
Compression Ratio Defined as the Ratio of the Compressed File Size to the Original File Size
Bold font indicates that a value has the best compression ratio among those tested for the given sequence. In MUHTL, a partition size of M = 4 was used since it gives better compression in most cases.
MUHTL, multiple unbalanced Huffman tree least frequent base; UHTL, unbalanced Huffman tree least frequent base.
NUHT tackles greediness by picking the 2-mer that is optimal, in the sense that by encoding it, the total of the 2-mer is maximized and the total of the base 4 is minimized. When this occurs, the 2-mer has a compression ratio impact of 18.75% and the base 4 has compression ratio impact of 37.5%, thereby contributing to a better overall compression ratio. When adding the impact of 2-mer and base 4, NUHT always has a lower compression ratio than UHT (Table 5).
The 2-mer and Base 4 Totals While Applying Nongreedy Unbalanced Huffman Tree and Unbalanced Huffman Tree Algorithms
The total is calculated as follows: total = 2-mer × 18.75% + base4 × 37.5%. Choosing 2-Mer and Base 4 results in a lower bits total which means better compression. When choosing both of them in a greedy way, it results in higher bit total.
For the special-purpose compressors in our comparisons (NUHT and UHT implementations), we tested an additional criterion: performance. A drawback of UHT implementations is the time they take to compress a relatively small DNA sequence. In its fastest implementation, the basic UHT algorithm requires around 30 seconds to compress a DNA sequence of around 4 Mb, and the time required increases with the file size. For example, a file size of around 40 Mb can take up to 5 minutes. Compared with UHT, NUHT encodes data seven times faster. NUHT takes 3, 5, 12, 43, and 44 seconds for compressing Cholerae, Abscessus, Saccharomyces_cerevi, Chr22, and Neurospora_crassa, respectively, while the basic UHT takes 31, 37, 90, 285, and 316 seconds for the same (Fig. 3). NUHT decompression times were also between three and seven times faster than those of UHT. This makes our tool preferable for larger files. Finally, we found that increase in file size makes UHT implementation unfeasible in terms of time.
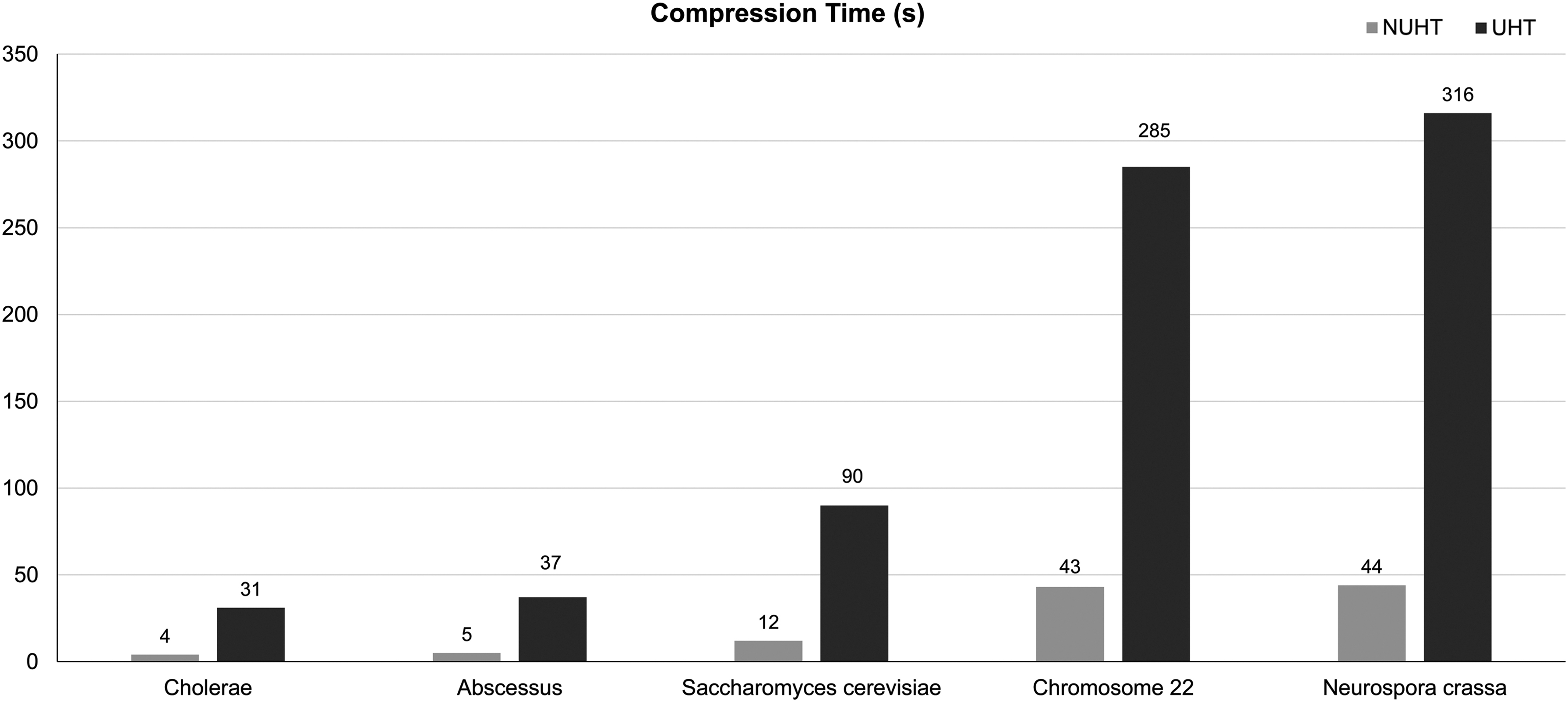
Time spent to compress each sequence.
6. Conclusion
In this article, we proposed a nongreedy Huffman tree-based compressor that surpasses other tools that use Huffman trees in terms of either the compression ratio or both speed and compression ratio. Our tool takes advantage of the 2-mer since it has the highest ratio impact factor. Our tool restricts the encoding process to one 2-mer along with four bases. The algorithm chooses the optimal 2-mer that leads to minimizing the base 4 count to build the optimal Huffman tree. This has shown a significant improvement over the greedy Huffman tree used in other tools. We compared our tool with gzip, bzip2, and UHT. For all data used in our comparison, our tool provided a better compression ratio and, when compared with UHT, offered significant improvement in speed. Our tool is freely available in binary files for several platforms at https://github.uconn.edu/sya12005/Non-Greedy-Unbalanced-Huffman-Tree-Compressor-for-single-and-multi-fasta-files
Authors' Contributions
S.A. initiated the research, developed and implemented the algorithms, and conducted the experimental study and analysis. C.H.H. directed the research.
Footnotes
Author Disclosure Statement
The authors declare they have no competing financial interests.
Funding Information
No funding was received.