
Abstract
Colorectal cancer (CRC) is a heterogeneous disease with distinct molecular properties. Tremendous works for CRC molecular subtyping are mainly based on gene expression profiling, which cannot capture the complementary information from other data types. Based on the classical multiomics data integration method similarity network fusion (SNF), which, however, suffers the trivial parameters setting, we developed local scaling SNF (Ls-SNF) that employs the local scaling method to construct patient affinity before network fusion. Local scaling infers the self-tuning of sample-to-sample distance and can eliminate the scaling problem. We have demonstrated the effectiveness of Ls-SNF on other cancer molecular subtyping in our previous study. In this study Ls-SNF applied in CRC molecular subtyping shows clear integrated patterns of gene expression, miRNA expression, and DNA methylation. Compared with the consensus molecular subtypes, subtypes identified by Ls-SNF achieved more significant association with clinical outcomes (p = 9.6 × 10−3, log-rank test). Certain mutations showed very significant enrichment in Ls-SNF subtypes, such as Class 3 were enriched for microsatellite instability (MSI) (p < 0.001), BRAF-mutant (p < 0.001), and CIMP high (p < 0.001). Ls-SNF subtypes also revealed better performance than some clinical risk factors in univariate and multivariate analyses (p = 0.002; p = 0.01).
1. Introduction
Colorectal cancer (CRC) is a heterogeneous disease with distinct molecular properties underlying diverse clinical outcomes. Recent technologies, such as next-generation sequencing techniques, increase the availability of multiomics molecular profiling, allowing us to dissect previously unknown functional diversity among various data layers. The accessibility of diverse omics data makes machine learning-based methods possible to capture the heterogeneity in cancer disease, such as the subtypes identification in breast cancer (Parker et al., 2009). Clustering of gene expression profiles has often been used to discover cancer subtypes. But only focusing on one data layer cannot obtain the valuable complementary information. Efficient integration methods should have the ability of generating a cluster assignment by combining several data types simultaneously. Typical example is the similarity network fusion (SNF) (Wang et al., 2014). SNF first constructs patient-to-patient similarity network based on correlation analysis and then iteratively fuses the patient similarity matrix by the nonlinear combination algorithm. The fused network can capture the common and complementary feature information from various data types, providing more insight into the observed patient similarities. Owing to the properties of fusion network, SNF not only obtains useful information from a small number of genes but also scales to a large set of available data measurements.
Tremendous effort has been dedicated to CRC subtyping in recent years. Schlicker et al. (2012) employed gene expression profiles of 62 CRCs as the discovery cohort to dissect the CRC heterogeneity based on non-negative matrix factorization. They identified two main subtypes (types 1 and 2) that were further split into five subtypes in the second iteration.
The two finding subtypes then were successfully validated on other 15 public gene expression data set. Marisa et al. (2013) performed unsupervised consensus hierarchical clustering on gene expression data from a discovery subset of 443 CRC samples and identified six molecular subtypes. These subtypes were associated with distinct molecular characteristics, molecular alterations, and specific enrichments of supervised gene expression signatures. Their findings were further validated using the remaining 123 samples and eight other independent public data sets for a total of 1058 CRC patients. Using gene expression profiles of 1290 CRC tumors, Sadanandam et al. (2013) identified six clinically relevant subtypes that were associated with distinct cell types in normal colon crypt by calculating the similarities between gene expression profiles and previous gene signatures using nearest template prediction algorithms (Hoshida, 2010). De Sousa E. Melo et al. (2013) categorized 90 stage II patients into three main colon cancer subtypes (CCSs) using unsupervised consensus-based clustering technique. The subtypes CCS1 and CCS2 were consistent with chromosomal instability (CIN) and microsatellite instability (MSI) subgroups, and CCS3 was newly identified subtype with worst prognosis. Their findings were then validated on six independent data sets and cell lines, including >1100 CRC patients.
The aforementioned CRC molecular subtyping strategies are based on gene expression profiling that can provide valuable comprehension of tumor biology in CRC patients. But integrating different data types can take advantage of the complementarity from different genomic measurements. Therefore, we aim to explore the multiomics data integration on molecular subtyping of CRC. In our previous study (Duan and Wang, 2018), we have demonstrated that local scaling SNF (Ls-SNF) outperformed the classical fusion method-SNF. Our approach incorporates local scaling (Zelnik-Manor and Perona, 2005) method to construct the affinity similarity network that allows the self-tuning of the patient-to-patient distance and can eliminate the scaling problem. In this study we applied Ls-SNF to CRC molecular subtyping. Comparing with the consensus molecular subtypes (CMS), CRC subtypes identified by our Ls-SNF are more molecularly distinct and clinically relevant.
2. Materials and Methods
2.1. Patients series
In this study we used two different CRC patient series with a total of 879 patients. The Cancer Genome Atlas (TCGA) (Zhang et al., 2014) CRC patients with multiomics data (N = 320) were downloaded, including gene expression, miRNA expression, and DNA methylation. Gene expression data were measured on Ilumminia HiSeq 2000 platform. The miRNASeq data were produced by Illumina HiSeq 2000 platform. For DNA methylation, the JHU-USC-Human-Methylation-450 platform was used. The Marisa et al. data set CIT cohort (Marisa et al., 2013) (GSE39582) comprises 559 CRC patients with clinical information and were measured on microarray platform (Affymetrix Human Genome U133 Plus 2.0).
2.2. Local scaling SNF
SNF uses a graph
Here
Local scaling: The parameters settings in SNF seem to be ambiguous and may fail to achieve good performance when data involve multiple scales. Instead, we propose to set local scaling parameters
This definition allows the self-tuning of point-to-point distances surrounding points i and j. According to the local statistics,
After the similarity affinity construction, we performed the similar steps to normalize and fuse the affinity matrix. The normalization is as follows:
Then, K nearest neighbors were employed to measure local affinity.
where
v are the data types. After t steps iteration, the final fused graph matrix is computed as
After obtaining the fused graph P, we can use classical clustering algorithms, such as spectral clustering to assign label to each patient. Comparing with the CMS, our Ls-SNF identified cancer subtypes that are much more molecularly distinct and clinically relevant.
2.3. Estimating the optimal clustering number
To select the optimal clustering number in fusion network, we analyzed the eigenvectors of the affinity matrix according to Zelnik-Manor and Perona (2005). Given communities C, we aim at finding an indicator matrix
We can define the following cost function to be minimized:
The optimal number of clusters is one of the communities that results in the largest drop in the value of
2.4. Statistical analysis
Statistical analyses were performed using R (version 3.4.3, www.r-project.org). Survival analyses were performed using the Kaplan–Meier method and statistical significance was evaluated using log-rank test from “survival package.” Univariate and multivariate Cox proportional hazards regression models were built using “coxph” function in “survival” package and hazard ratios were calculated using function “hazard.ratio” in “survcomp” package. p-Value <0.05 was considered significant for all tests. Gene set enrichment analysis was performed using “HTSanalyzeR” package.
3. Results and Discussion
We carried out the molecular subtyping analysis of CRC using Ls-SNF (Fig. 1). In preprocessing step, genes with high variability were kept for downstream analysis by median absolute deviation (MAD >0.5) (Howell, 2014). We then constructed local scaling affinity in each preprocessed data type. Network fusion was iteratively performed on the affinities to integrate the three data types. After network fusion, we implemented spectral clustering to assign a label to each patient (Fig. 1) since spectral clustering has the advantage of capturing the global graph structure. We estimated the optimal clustering number with K = 4 by analyzing the eigenvectors of the fusion network (Zelnik-Manor and Perona, 2005) (Supplementary Fig. S1).

Ls-SNF for colorectal cancer molecular subtyping pipeline. We take as input multiomics data of TCGA colorectal cancer (N = 320). Ls-SNF is then capable of constructing patient-to-patient affinity matrix by the local scaling method. Affinity matrices are fused into one graph by nonlinear combination. Subsequently, clustering is performed on the fusion graph, followed by the molecular and clinical characterization. Ls-SNF, local scaling similarity network fusion; TCGA, The Cancer Genome Atlas.
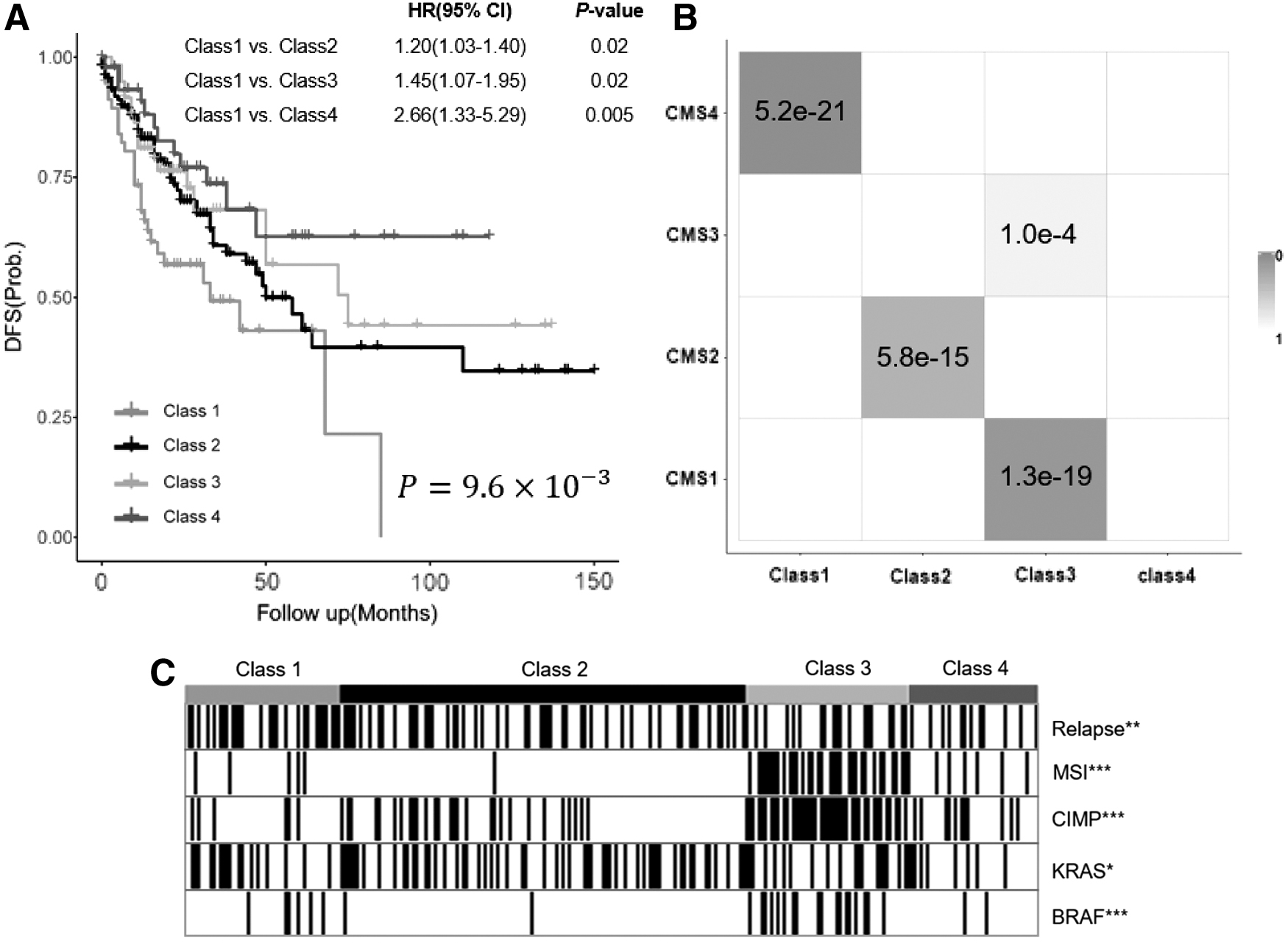
We first evaluated the clustering performance by examining the cluster pattern on the fusion graph. The fused graph reveals a clear picture of clustering in colorectal patients, demonstrated by the tightness of connectivity within clusters (Fig. 2A) and relatively few edges between clusters (Fig. 2B). To study the clinical relevance of the Ls-SNF subtypes, we performed survival analysis. Disease-free survival (DFS) was used to indicate the clinical association of the clustering results. The difference between the population survival curves can be indicated by the log-rank test p-value. Benchmarking against the CMSs (Supplementary Fig. S2), subtypes identified by Ls-SNF achieved more significant association with clinical outcomes (p = 9.6 × 10−3, log-rank test) (Fig. 3A). To determine whether the Ls-SNF subtypes differed in outcome, we performed Cox proportional hazards analysis. Class 1 subtype had the worst DFS.
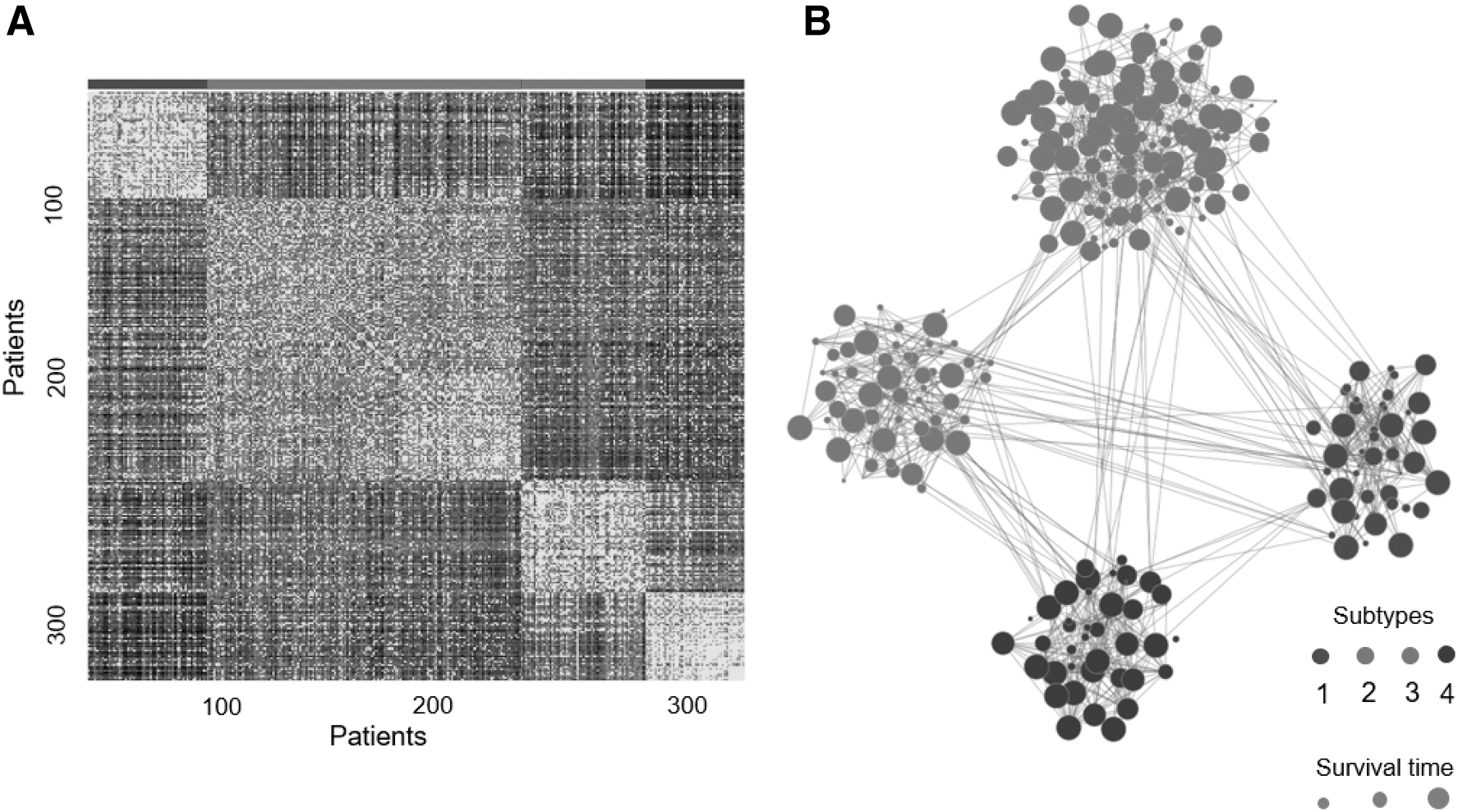
Patient similarity in Ls-SNF fusion graph. Patient-to-Patient similarities for 320 colorectal cancer patients visualized by fusion similarities matrix
Ls-SNF identified four CRC subtypes.
To explore the association between Ls-SNF subtypes and molecular characteristics, we performed molecular characteristics analysis based on the Ls-SNF subtypes. Certain mutations showed very significant enrichment in Ls-SNF subtypes. Class 3 were enriched for MSI, BRAF-mutant, and CIMP high, indicating that this subtype demarcates the well-characterized MSI/CIMP+ subset of CRC. Class 2 showed a high level of KRAS mutations. Class 1 had the most frequent relapse and showed a significantly poor prognosis (Fig. 3C). To measure the consistency between our Ls-SNF subtypes and the CMSs, hypergeometric test was used for pairwise comparisons. The four subtypes identified by Ls-SNF achieved a high concordance with the four CMSs (Fig. 3B), as indicated by significant p-values.
To compare the prognostic significance of Ls-SNF subtypes with other risk factors, we performed univariate and multivariate Cox regression analyses. From univariate and multivariate analyses, besides TNM staging, Ls-SNF subtypes showed significant association with patient survival (Table 1). To examine the biological pathways characterized in each subtype. We performed gene enrichment analysis based on known signatures. Class 1 tumors displayed significant upregulation of genes associated with epithelial–mesenchymal transformation and activation of transforming growth factor-β signaling, matrix remodeling pathways (Fig. 3A). In addition, Class 1 samples showed a gene expression profile compatible with stromal infiltration (Fig. 4C). Enrichment of multiple metabolism signatures was found in Class 2 epithelial CRCs, which was consistent with KRAS-activating mutations and has been treated as inducing significant metabolic adaptation (Brunelli et al., 2014) (Fig. 4D). Class 3 tumors are characterized by gene expression associated with diffuse immune infiltration, largely consisting of TH1 and cytotoxic T cells, and in line with strong activation of immune evasion pathways, which are emerging features of MSI CRC (Llosa et al., 2015) (Fig. 4C–E). Class 4 subtype showed epithelial differentiation and significant upregulation of WNT and MYC, both of which are often associated with CRC carcinogenesis (Fig. 4A).

Gene set mRNA enrichment analysis revealing signatures of interest in CRC, including canonical pathways, immune signatures, immune and stromal cell admixture in CRC patients, and metabolic pathways.
Univariate and Multivariate Analysis
CI, confidence interval; dMMR, defective DNA mismatch repair; HR, hazards ratio; MMR, mismatch repair; MSI, microsatellite instability; MUT, mutation; WT, wild type.
Next, using the TCGA gene expression data set for training, we validated the identified subtypes on GSE39582 (n = 599) data set. We applied the random forest to train the classifier and taken as input the differential genes in the four classes. Survival analysis based on our validation subtypes showed significant association with relapse-free survival (Supplementary Fig. S3).
4. Conclusions
Integrating multiple genome-scale data can capture complementary information from various data layers and helps understand biological processes. The classical method SNF, which is based on nonlinear network combination, can fuse different types of genome-wide data into one representative graph. SNF has been successfully applied to the identification of cancer subtypes. In this article, based on SNF, we propose to construct affinity matrix using local scaling method before network fusion and apply it to molecular subtyping of CRC. This affinity construction method requires less parameter setting and eliminates the scaling problem, resulting in strong robustness to reveal intrinsic molecular properties associated with different cancer subtypes. Compared with the classical CMS, the subtypes identified by Ls-SNF have a stronger association with survival. Gene set enrichment analysis indicates our Ls-SNF subtypes show distinct molecular characteristics and clinical associations. We hope that the improvement in cancer subtyping strategies will take us closer to clinical translation for patient treatments and the design of new targeted drugs.
Footnotes
Author Contributions
F.G. and X.J.W. conceived and designed the project; X.D. and F.G. collected the data. X.D. analyzed and interpreted the data; X.D. and F.G. drafted the article; K.J.W., J.K., and C.Y.F. reviewed the article; funding acquisition was by X.J.W. All authors read and approved the final article.
Author Disclosure Statement
The authors declare they have no competing financial interests.
Funding Information
This study was supported by National Key Clinical Discipline, Science and Technology Planning Project of Guangdong Province (No. 2017B020226001), Guangdong Natural Science Foundation (No. 2017A030310517), and the Fundamental Research Funds for the Central Universities (No. 17ykpy66).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.