
Abstract
To provide systematic insight into the composition and expression of transfer RNA (tRNA) derivatives transcriptome in colorectal cancer (CRC). tRNA derivatives expression profiles in three pairs of CRC and adjacent normal colon tissues were performed by tRNA-derived small RNA fragments (tRFs) and tRNA halves (tiRNA) sequencing, and microarray data of transcriptomes from CRC and paired controls were retrieved from Gene Expression Omnibus database. The differentially expressed tRFs and tiRNAs and differentially expressed genes between CRC and paired normal samples were screened. The functional regulations between tRF and tiRNA and gene were identified. A total of 60 upregulated and 48 downregulated tRNA derivatives and 7373 upregulated and 12,138 downregulated messenger RNA (mRNA) were identified. The tRF and tiRNA-gene regulatory modules were constructed by analyzing computational tRF and tiRNA-target predictions and inverse expression relationships between tRF and tiRNAs and mRNA. Gene ontology and Kyoto Encyclopedia of Genes and Genomes pathway annotation showed that the function of targets of tiRNA-Tyr-GTA was mainly enriched in negative regulation of epithelial cell apoptotic process and peroxisome proliferator activated-receptors (PPAR) signaling pathway. Cellular response to monoamine stimulus and inflammatory bowel disease was enriched in function of tiRNA-Val-CAC. Two functions, including negative regulation of c-Jun N-terminal kinase (JNK) cascade and choline metabolism in cancer, were enriched in tRF-Gln-CTG. The function of mesenchymal to epithelial transition was enriched in tRF-Leu-TAG. For the first time to our knowledge, our study provided a landscape of tRNA derivatives expression profiles in CRC. Further tRF and tiRNA-gene regulatory modules construction explored the potential functions related to these tRNA derivatives in the pathogenesis of CRC.
1. Introduction
Colorectal cancer (CRC) is the fourth most common cancer in Asia and is among the most frequent causes of mortality worldwide (Ferlay et al., 2015; Park et al., 2016). The estimate CRC incidence is expected to increase by 60% to >2.2 million new cases and 1.1 million deaths by 2030 (Arnold et al., 2017). Although the overall survival of CRC patients had markedly improved within the last decade, to date, the median progression-free survival was only 9.3 months and the median overall survival was 20.5 months in stage IV CRC patients (Rothenberg et al., 2008). Despite the effectiveness of surgical resection, radiation, and chemotherapy in the treatment of nonstage IV CRC, the disease may eventually progress in almost 25% patients (Abulafi and Williams, 1994). Therefore, searching for biomarkers and effective molecular targets for CRC is essential.
Transfer RNAs (tRNAs), which are housekeeping products, are a fundamental component of the translation machinery by acting as adapters in protein synthesis (Goodarzi et al., 2016). Apart from their canonical function, tRNAs were demonstrated to play influential roles in regulating cell proliferation and cancer progression by altering the expression of specific transcripts and proteins (Goodarzi et al., 2016). tRNA degradation debris, namely tRNA derivatives or tRNA-derived fragments, also had a vital role in cancer (Goodarzi et al., 2016). Based on size, tRNA derivatives were divided into two subtypes: tRNA halves (tiRNAs) and tRNA-derived small RNA fragments (tRFs).
tiRNAs, including 5′ tiRNAs and 3′ tiRNAs with sizes of 30–35 nt, were produced by specific endonucleolytic cleavage at the anticodon loop of the mature tRNA (Lee et al., 2009). tRFs are relatively smaller RNAs (14–30 nt) formed by nucleases such as Dicer and RNase Z at the 5′ or 3′ end of mature or precursor tRNAs and were classified into at least three types, including the 5′-tRF, 3′-tRF, and tRF-1 series (Shen et al., 2018). These tRNA derivatives can repress messenger RNA (mRNA) targets in micro RNA (miRNA)-like manner, thereby regulating the proliferation of cancer cells (Haussecker et al., 2010). tRF5-Glu, a 5′- tRF, binds directly to a site in the 3′-untranslated region (3′-UTR) of the BCAR3 mRNA, thereby downregulating its expression. Inhibiting BCAR3 expression was reported to suppress ovarian cancer cell proliferation. Furthermore, mimics of tRF5-Glu were found to inhibit the proliferation of ovarian cancer cells (Zhou et al., 2017). Another 5′- tRF, tRF-Leu-CAG, was demonstrated to be involved in regulating AURKA. Its knockdown resulted in repression of AURKA and suppression of human nonsmall cell lung cancer cell proliferation (Shao et al., 2017). Taken together, these researches strongly revealed a functional role of tRNA derivatives in tumorigenesis.
To date, little is known about the relationship between tRNA derivatives and CRC. Yet, studies providing systematic insight into the composition and expression of the tiRNAs and tRFs transcriptome in CRC are still missing. Therefore, in this study, we screened the tRNA derivative profiles of CRC patients using high-throughput tRF and tiRNA sequencing. Then, we utilized an integrative strategy to construct functional tRF/tiRNA-gene regulatory networks by combining the reverse expression relationships between tRF/tiRNAs and targets and computational predictions. Our study may provide clues to better understand the potential roles of tRNA derivatives in the pathogenesis of CRC.
2. Methods
2.1. Study samples and data sets
A total of three pairs of CRC and adjacent normal colon tissues were collected from patients at the Department of Colorectal Surgery, Union Hospital, Fujian Medical University, in June 2018. The diagnosis of CRC was independently confirmed by two pathologists. The patients did not receive any radiotherapy or chemotherapy before surgical resection. All tissue samples were snap-frozen and stored in liquid nitrogen. This study was approved by the ethics committee of Union Hospital, Fujian Medical University, and written informed consent was obtained from all patients.
Microarray data of transcriptomes from CRC and paired controls were retrieved from the Gene Expression Omnibus (GEO) database (www.ncbi.nlm.nih.gov/geo). The basic information of these data sets is displayed in Table 1.
Basic Information of Microarray Data Used in This Study
CRC, colorectal cancer.
Schematic overview of the proposed approach in this study is shown in Figure 1.

Schematic overview of the proposed approach in this study. CRC, colorectal cancer; DEGs, differentially expressed genes; DETs, differentially expressed tRFs and tiRNAs; mRNA, messenger RNA; tiRNAs, tRNAs halves; tRF, tRNA-derived small RNA fragments; tRNA, transfer RNA.
2.2. RNA extraction and quality control
Total RNA was isolated from tissue samples using TRIzol reagent (Thermo Fisher Scientific, Inc., Carlsbad, CA). The quality of the RNA samples was determined by OD 260/280 assessment. Agarose gel electrophoresis was used to check the integrality of total RNA samples, and the samples were quantified on the NanoDrop ND-1000 instrument (Thermo Fisher Scientific, Waltham, MA).
2.3. Library preparation and tRF and tiRNA sequencing
Total RNA samples were first pretreated as following to remove some RNA modifications that interfere with small RNA-seq library construction: 3′-aminoacyl (charged) deacylation to 3′-OH for 3′-adaptor ligation, 3′-cP (2′, 3′-cyclic phosphate) removal to 3′-OH for 3′-adaptor ligation, 5′-OH (hydroxyl group) phosphorylation to 5′-P for 5′-adaptor ligation, and m1A and m3C demethylation for efficient reverse transcription. Pretreated total RNA of each sample was taken for tRF and tiRNA-seq library preparation. Library preparation procedures included (1) 3′-adapter ligation, (2) 5′-adapter ligation, (3) complementary DNA synthesis, (4) polymerase chain reaction (PCR) amplification, and (5) size selection of ∼134–160 bp PCR amplified fragments (corresponding to ∼14–40 nt small RNAs). The completed libraries were quantified by Agilent 2100 Bioanalyzer. Libraries were mixed in equal amounts according to the quantification results and used for sequencing on the instrument.
The DNA fragments in well-mixed libraries were denatured with 0.1 M NaOH to generate single-stranded DNA molecules and loaded onto the reagent cartridge at 1.8 pM concentration. The sequencing run was performed on NextSeq system using NextSeq 500/550 V2 Kit (#FC-404-2005; Illumina) according to the manufacturer's instructions. Sequencing was carried out by running 50 cycles.
Sequencing quality was examined by FastQC and trimmed reads were aligned allowing for one mismatch only with the mature tRNA sequences, then reads that did not map were aligned allowing for one mismatch only with precursor tRNA sequences with Bowtie software (Langmead et al., 2009). The expression profiling of tRF and tiRNA was calculated based on counts of reads mapped. The abundance of tRF and tiRNA was evaluated using their sequencing counts and was normalized as counts per million of total aligned reads (CPM).
2.4. Identification of tRF and tiRNAs and genes (mRNA) associated with CRC
The differentially expressed tRFs and tiRNAs (DETs) were screened based on the count value with R package edgeR (Robinson et al., 2010). Principal component analysis (PCA), pie plots, Venn plots, hierarchical clustering, scatter plots, and volcano plots were performed in R 3.5.1 or Perl environment.
GEO2R was performed to identify differentially expressed genes (DEGs) between CRC and paired normal samples. GEO2R is an interactive webtool that allows gene expression analysis of microarray data sets using Limma R packages (Davis and Meltzer, 2007). p-Value <0.05 and fold change >2 or <0.5 were set as the cutoff criteria. To incorporate more candidate genes, the union set of DEGs in three data sets was used.
2.5. tRF and tiRNA target prediction
To explore the potential regulations between tRF and tiRNA and gene in CRC, we performed both the computational target prediction analysis and parallel mRNA and tRF and tiRNA expression analysis. The functional regulations were identified by (1) both DETs and DEGs associated with CRC, (2) computational predictions at the sequence level, and (3) inverse regulation between DETs and DEGs.
The computational prediction of regulatory relationships between tRF and tiRNAs and targets was determined using TargetScan (Agarwal et al., 2015; www.targetscan.org) and miRanda (Betel et al., 2010; www.microrna.org). In this study, we restricted our search to minimum tRF and tiRNA seed length of seven nucleotides and binding sites on the 3′-UTR of the target gene. The putative target gene binding sites were identified by searching for the presence of conserved 8 mer, 7 mer, and 6 mer sites that match the seed region of each DET. Inverse expression relationships were then identified between upregulated/downregulated DETs and downregulated/upregulated DEGs. The common target genes in both computational prediction analysis and parallel mRNA and tRF and tiRNA expression analysis were regarded as “real” target genes. Finally, tRF and tiRNA-gene regulatory module analysis was conducted, and the modules were assembled and visualized using Cytoscape 3.6.1 software (Shannon et al., 2003).
2.6. Functional enrichment analysis
To associate tRF and tiRNA with specific biological function, functional enrichment analysis was performed for the target genes of DETs based on gene ontology (GO) and based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways annotation. All the genes included in each tRF and tiRNA-gene regulatory module were used in the functional enrichment analysis with the Cytoscape 3.6.1 plug-in software, ClueGO v2.5.3 (Bindea et al., 2009). The significance value was set as p-value <0.05.
2.7. Statistics
Continuous variables were compared using Student's t-test. All statistical analyses were performed using the SPSS software (version 17 SPSS, Inc., Chicago, IL), GraphPad prism 7 and R (version 3.4.1). A p-value <0.05 was taken as the level of significance.
3. Results
3.1. Overview of tRF and tiRNA profiles
A total of 327 tRNA derivatives were identified by tRF and tiRNA-seq in this study, and 261 of them were identified as new tRNA derivatives that had never been annotated in the tRFdb database (Kumar et al., 2014; Fig. 2A). Of the tRNA derivatives identified, 250 tRNA derivatives of the CPM values, which were >20, were selected for further analysis. tRNA isodecoders sharing the same anticodon might have different body sequence, and belong to different tRF and tiRNA subtypes.
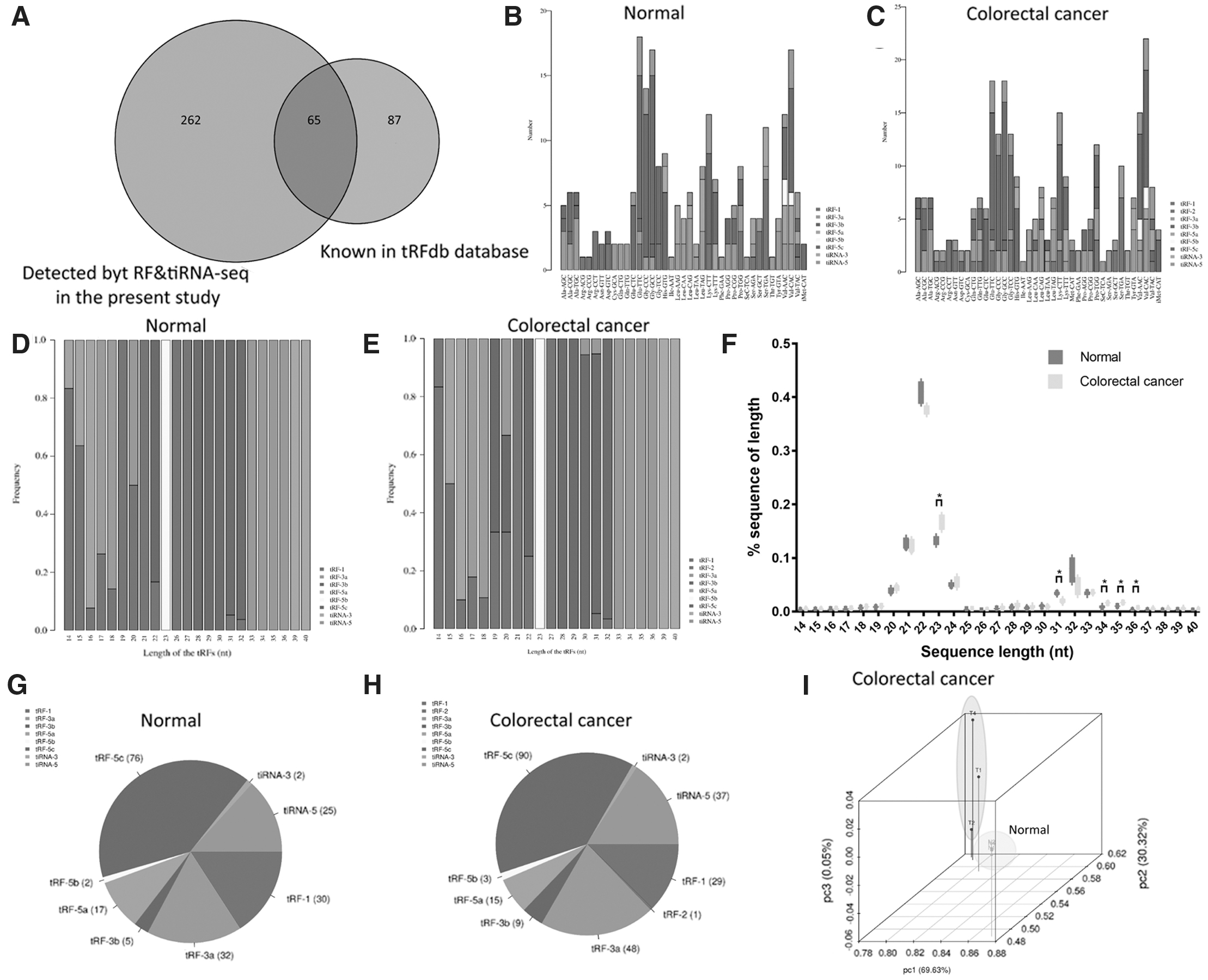
Overview of tRF and tiRNA profiles.
The stacked plot showed that the number of tRF and tiRNA subtypes derived from the same anticodon tRNA (Fig. 2B, C). The frequency of subtypes against the length of the tRFs and tiRNA is shown in Figure 2D and E. tiRNA-3 has the longest length of 39–40 nt, followed by tiRNA-5 with a length of 33–36 nt in both CRC and paired normal tissue. The length of tRFs varied, between 14 and 32 nt. All tRF5-b were 23 nt long, and the length of tRF3-a was within 17–18 nt in both CRC and normal colon mucosa. All tRF3-b were 22 nt long in normal colon mucosa, but the length of tRF3-b could be 19, 20, and 22 nt in CRC. The length distribution of tRFs and tiRNAs in CRC differed characteristically from those of tRFs and tiRNAs in normal tissue (Fig. 2F). The lengths of 23 nt (refers to tRF-5b) and 34–36 nt (refers to tiRNA-5) were more frequent in CRC samples, whereas the length of 31 nt (refers to tRF-5c or tRF-1) was more frequent in normal samples. The distribution of tRF and tiRNA subtypes in CRC and normal colon mucosa is shown in Figure 2G and H. tRF5-c was the most abundant isoform, followed by tRF3-a. PCA of strongly expressed tRFs and tiRNAs showed a complete separation of CRC and normal controls (Fig. 2I).
3.2. Identification of tRF and tiRNA and genes (mRNA) associated with CRC
DET and DEG analyses were performed between CRC and paired normal samples. A total of 60 upregulated and 48 downregulated tRNA derivatives and 7373 upregulated and 12,138 downregulated mRNA satisfied the significant threshold (Fig. 3A–C). The top three up- and downregulated tRNA derivatives are listed in Table 2 according to fold change. Then, these six most differentially expressed tRNA derivatives, including two tiRNAs and four tRFs, were selected to construct tRF and tiRNA-gene regulatory modules. Among them, tiRNA-Val-CAC, tiRNA-Tyr-GTA, and tRF-Gln-CTG were upregulated, and tRF-Val-AAC, tRF-Leu-TAG, and tRF-Leu-AAG were downregulated in CRC samples compared with paired normal samples (Fig. 3D).

Identification of tRF and tiRNAs associated with CRC.
Top Six Differentially Expressed tRFs and tiRNAs in Colorectal Cancer
tiRNAs, tRNAs halves; tRF, tRNA-derived small RNA fragments; tRNA, transfer RNA.
3.3. tRF and tiRNA target prediction
The tRF and tiRNA-gene regulatory modules were constructed using two independent and complementary types of information: computational target predictions and inverse expression relationships. Because tRNA derivatives suppress the translation of target genes and regulate the expression of target genes in a manner similar to miRNAs, we investigated genes potentially associated with tRNA derivatives by computational prediction of binding sites between tRNA derivatives and the 3′-UTR of the target gene. For example, tRF-Leu-AAG is predicted to harbor WIPI2, CSRNP2, CACYBP, FAM13A, AURKA, and ADGRF5 with seed sequence matching type of 8 mer (Fig. 3E). Then, 151 putative target genes of selected downregulated DGTs were real upregulated DGEs, and 882 putative target genes of selected upregulated DGTs were real downregulated DGEs. In addition, a tRF and tiRNA-gene regulatory module network was constructed (Fig. 4).
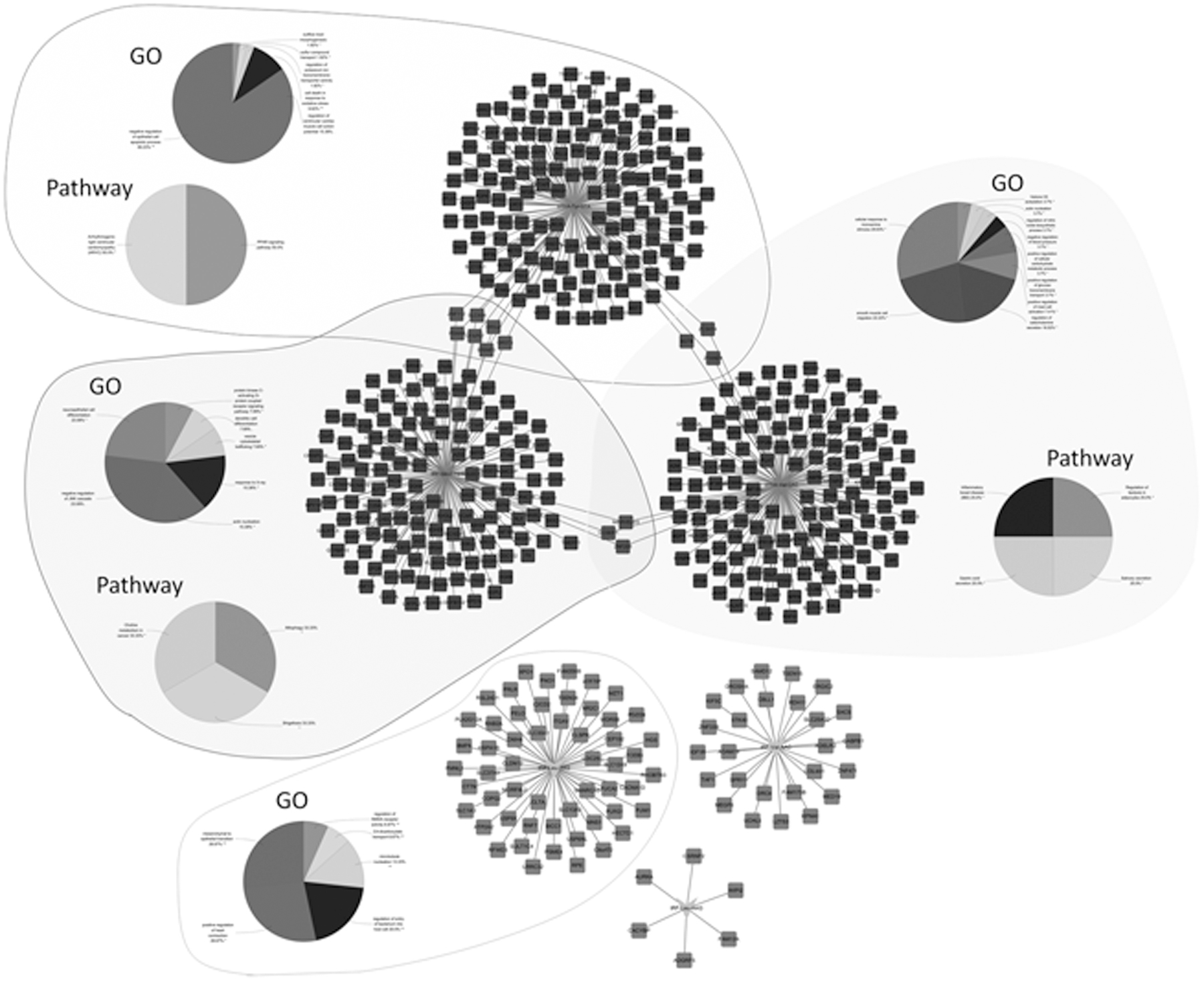
tRF and tiRNA-gene regulatory module network construction and functional enrichment analysis. Black represents upregulated tRF and tiRNAs or mRNAs, and gray represents downregulated tRF and tiRNAs or mRNAs. The regulatory modules were constructed by integrated analysis of computational target predictions and inverse expression relationships. GO, gene ontology.
3.4. Functional enrichment analysis
GO and KEGG pathway annotation revealed that the function of host genes of tiRNA-Tyr-GTA was mainly enriched in negative regulation of epithelial cell apoptotic process and peroxisome proliferator activated-receptors (PPAR) signaling pathway. Two functions, including cellular response to monoamine stimulus and inflammatory bowel disease (IBD), were enriched in tiRNA-Val-CAC. Two functions, including negative regulation of c-Jun N-terminal kinase (JNK) cascade and choline metabolism in cancer, were enriched in tRF-Gln-CTG. The function of mesenchymal to epithelial transition was enriched in tRF-Leu-TAG. Owing to relatively small number of target genes, no significant biological function and pathway were enriched in tRF-Leu-AAG and tRF-Val-AAC (Fig. 4).
4. Discussion
tRNAs, as classic ncRNAs, were traditionally considered to act as an indispensable component in protein synthesis and were generally neglected. However, with the use of powerful high-throughput sequencing technologies, more comprehensive insights into tRNA transcriptomes had become feasible, and the number of studies on tRNA has increased gradually in recent years (Pavon-Eternod et al., 2009). The tRNA overexpression, which coordinates with the properties of cognate amino acids, may improve the translational efficiency of cancer-related genes (Pavon-Eternod et al., 2009). tRF and tiRNA analysis system based on Illumina NextSeq 500 system, used in this study, is a potent tool for the analysis of tRNA derivatives. A total of 327 tRNA derivatives, which are specific cleavage of tRNAs by specific nucleases, were identified by tRF and tiRNA-Seq in this study. In addition, we identified 261 novel tRNA derivatives from the RNA-Seq data. These new tRNA derivatives are especially interesting for further research because of the lack of information about them in current databases (Kumar et al., 2014).
Moreover, since there is still a lack of global repository for sequences of tRNA derivatives, further annotation of these new tRNA derivatives is needed. In addition, the length of tiRNAs in this study was longer than that of tRFs. Although the length of tRFs varied between 14 and 32 nt, each tRF subtype had a relatively fixed length range. This ultrahigh enrichment feature of length distributions elucidated that tRNA derivatives, including tRFs and tiRNAs, were not random degradation products but the result of accurate cleavage modulations (Li et al., 2016). Recent findings demonstrated that different types of tRFs and tiRNAs were generated by different specific ribonucleases, such as angiogenin, dicer, or ribonuclease (Olvedy et al., 2016).
The modulating function of tRNA derivatives in diverse biological processes was gradually explored in recent years (Telonis and Rigoutsos, 2017). Huang et al. (2017) reported that tRF/miR-1280, a 17-bp fragment derived from tRNA-Leu, inhibited Notch signaling pathways by targeting Notch ligand JAG2, and decreased cancer stem-like cells in CRC progression. However, there is still a lack of systematic study investigating the involvement of tRNA derivatives in CRC progression by tiRNAs and tRFs transcriptome analysis. In this study, we intended to improve the understanding of tRNA derivatives expression pattern in CRC. By comparing the length distribution of CRC and normal tissues, we found that the length of 23 nt (matches with tRF-5b in this study) and 34–36 nt (matches with tiRNA-5) was more frequent in CRC samples, whereas the length of 31 nt (matches with tRF-5c or tRF-1) was more frequent in normal samples. Further DMT analysis demonstrated that two tiRNA-5s (tiRNA-Val-CAC and tiRNA-Tyr-GTA) were the most upregulated tRNA derivatives in CRC compared with normal tissues, and one tRF-5b (tRF-Val-CAC) and one tRF-1 (tRF-Val-CAC) were highly expressed in CRC and normal tissue, respectively (Fig. 3; Table 2). These biomolecules might offer potential diagnostic biomarkers of CRC for further studies. Although the PCA of strongly expressed DMTs could completely distinguish CRC from normal controls, the sample size in this study was too small. Moreover, experimental validation of the occurrence of these biomolecules in cell and tissue levels by quantitative real-time PCR is needed in our future study.
This study performed an integrating analysis and then constructed tRF and tiRNA-gene regulatory modules based on both the computationally predicted target interactions and parallel mRNA and tRF and tiRNA expression levels. The disease-associated mutations contained the disease-relevant genes and targeting regulators were thought to have the potential for identifying drug targets and biomarkers for complex diseases (Barabási et al., 2011). By functional enrichment analysis, we found that the function of host genes of tiRNA-Tyr-GTA was mainly enriched in negative regulation of epithelial cell apoptotic process and PPAR signaling pathway. It is known that the apoptotic signals contribute to safeguarding the genomic integrity while the defect in apoptosis may promote carcinogenesis (Hassan et al., 2014). PPAR is a ligand-activated transcription factor that plays a critical role in regulating cancer progression, and its role of tumorigenesis was first identified in CRC (You et al., 2015).
The function of another upregulated tiRNA, tiRNA-Val-CAC, might be involved in the cellular response to monoamine stimulus and IBD in this study. Interestingly, monoamine oxidase inhibitor, an antidepressant, was associated with an increased incidence of CRC in a population-based nested case–control study (Lee et al., 2017). In contrast, depression was reported to increase the risk of IBD (Frolkis et al., 2018), and it is widely known that patients with IBD are at increased risk of CRC, principally resulting from the proneoplastic effects of chronic intestinal inflammation (Croog and Itzkowitz, 2004). The complicated role of tiRNA-Val-CAC in the relationship among depression, IBD, and CRC needs further research. Two functions, including negative regulation of JNK cascade and choline metabolism in cancer, were enriched in tRF-Gln-CTG. The inhibition of JNK cascade was reported to reduce the migration potential of human colon carcinoma cells in vitro (Jemaa et al., 2018). Previous research based on tissue metabolic profiling of lymph node metastasis showed that disturbance of choline metabolism was affected in CRC, which suggested the promising application of these metabolites in clinical therapy (Zhang et al., 2016). Therefore, tRF-Gln-CTG might be involved in the metastasis of CRC by affecting their metabolism. In addition, the function of mesenchymal to epithelial transition, a dynamically dysregulated process involved in CRC metastasis process (Chou et al., 2016), was enriched in tRF-Leu-TAG. The specific roles remain to be further explored.
In conclusion, for the first time to our knowledge, our study revealed a landscape of tRNA derivatives expression profiles in CRC. The further tRF and tiRNA-gene regulatory modules construction based on target interactions and parallel mRNA and tRF and tiRNA expression levels explored the potential functions related to these tRNA derivatives, which might help us to understand the underlying pathogenesis of CRC better. In addition, these findings may provide potential diagnostic biomarkers and therapeutic targets for CRC. Further investigation is needed.
Footnotes
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This study was financially supported by the National Clinical Key Specialty Construction Project (General Surgery) of China (Grant No. 2012-649), National Natural Science Foundation of China (81902378), Young Scientist Foundation of Fujian Provincial Commission of Health and Family Planning (Grant No. 2017-1-39), and the Startup Fund for Scientific Research, Fujian Medical University (Grant Nos. 2016QH027 and 2017XQ1028), Joint Funds for the Innovation of Science and Technology, Fujian province (Grant No. 2017Y9038), Joint Funds for the Innovation of Science and Technology, Fujian province (Grant No. 2017Y9103), and Fujian Science and Technology Project (Grant No. 2016J01456).