
Abstract
AIDS is a syndrome caused by the HIV. During the progression of AIDS, a patient's immune system is weakened, which increases the patient's susceptibility to infections and diseases. Although antiretroviral drugs can effectively suppress HIV, the virus mutates very quickly and can become resistant to treatment. In addition, the virus can also become resistant to other treatments not currently being used through mutations, which is known in the clinical research community as cross-resistance. Since a single HIV strain can be resistant to multiple drugs, this problem is naturally represented as a multilabel classification problem. Given this multilabel relationship, traditional single-label classification methods often fail to effectively identify the drug resistances that may develop after a particular virus mutation. In this work, we propose a novel multilabel Robust Sample Specific Distance (RSSD) method to identify multiclass HIV drug resistance. Our method is novel in that it can illustrate the relative strength of the drug resistance of a reverse transcriptase (RT) sequence against a given drug nucleoside analog and learn the distance metrics for all the drug resistances. To learn the proposed RSSDs, we formulate a learning objective that maximizes the ratio of the summations of a number of ℓ1-norm distances, which is difficult to solve in general. To solve this optimization problem, we derive an efficient, nongreedy iterative algorithm with rigorously proved convergence. Our new method has been verified on a public HIV type 1 drug resistance data set with over 600 RT sequences and five nucleoside analogs. We compared our method against several state-of-the-art multilabel classification methods, and the experimental results have demonstrated the effectiveness of our proposed method.
Introduction
According to estimations by the World Health Organization, around 35 million people are suffering from the HIV. HIV is a serious virus that attacks cells in the human immune system. During the later stages of the virus it can critically weaken the immune system and increase the patient's susceptibility to serious infection and disease. Fortunately, with the advent of antiretroviral therapies, we have been able to stem the progression of HIV and extend the life span of individuals affected by the virus. Unfortunately, the high mutation rates of HIV type 1 (HIV-1) can produce viral strains that adapt very quickly to new drugs (Smyth et al., 2012). The mutation of HIV-1 during antiretroviral treatments can lead to a phenomenon called “cross-resistance” (Heider et al., 2013; Riemenschneider et al., 2016). Cross-resistance of HIV-1 occurs when the virus develops resistance against the drugs, which are currently being used in addition to other drugs that have not yet been used in the treatment of a particular patient. This can make treatment of HIV-1 significantly more difficult, because a collection of drugs may not be effective after the initial treatment regimen due to the cross-resistance phenomenon observed in HIV-1. To address this problem, it is important that we develop automatic methods that can associate genetic strains of HIV to their corresponding drug resistances. The success of this research has the potential to reduce health care costs and increase the quality of life of those suffering from HIV and AIDS.
Recently, experimental testing of viral resistance in patients has been widely used in research, as well as in clinical settings to gain insight into the ways in which the drug resistance evolves. For example, large-scale pharmacogenomic screens have been conducted to explore the relationships between drug resistances and genomic sequences (Rhee et al., 2003). Besides, many clinical trials have been performed to discover mutation rates of the genetic subtypes of HIV-1 and how they develop resistances against various drug treatments (Pennings, 2012). In addition to these experimental phenotypic studies, computational approaches that use various machine learning methods offer the possibility to predict drug resistance in HIV-1 using short sequence information of the viral genotype, such as the genetic sequence of the viral reverse transcriptase (RT). For example, Rhee et al. (2006) used five different machine learning methods, including decision trees, artificial neural networks, support-vector machines, least-square regression, and least-angle regression, to investigate drug resistance in HIV-1 based on the RT sequences. Besides, genotype and phenotype features of HIV-1 extracted from RT sequences have been studied to predict drug resistance (Hepler et al., 2014).
In addition, a Bayesian algorithm (Gönen and Margolin, 2014) that combines kernel-based nonlinear dimensionality reduction and binary classification has been proposed to predict drug susceptibility of HIV within a multitask learning (MTL) framework. A critical drawback of these existing studies lies in the fact that they routinely consider HIV-1 drug resistance prediction as a single-label classification problem. This approach has been recognized to be inappropriate since HIV strains can develop resistances against multiple drugs at once due to their high mutation rate (Heider et al., 2013; Riemenschneider et al., 2016). To tackle this difficulty, following Heider et al., 2013 in this article, we solve the problem of HIV-1 drug resistance prediction as a multilabel classification problem.
Multilabel classification is an emerging research topic in machine learning driven by the advances of modern technologies in recent years (Wang et al., 2009, 2010a–c, 2015). As a generalization of traditional single-label classification that requires every data sample to belong to one and only one class, multilabel classification relaxes this restriction and allows one data sample to belong to multiple different classes at the same time. As a result, the classes in single-label classification problems are mutually exclusive, while those in multilabel classification problems are interdependent on one another. Although the labeling relaxation in multilabel classification problems have brought a number of successes in a variety of real-world applications (Wang et al., 2009, 2010c, 2015), it also causes labeling ambiguity that inevitably complicates the problem (Wang et al., 2010a,b).
In the context of predicting drug resistance developed by HIV-1, some HIV strains can develop the capability to resist multiple drugs, including those currently being used and those that have not yet been applied in a clinical setting. As a result, it is often unclear how to utilize a data sample that belongs to multiple classes to train a classifier for a given class (Wang et al., 2010a,b). A simple strategy to solve this problem is to use such data samples as the training data for all the classes to which they belong (Wang et al., 2009, 2010a), which is equivalent to assume that every data sample contributes equally to all their belonging classes when we train multilabel classification model (Wang et al., 2010b). However, this is not always the case in many real-world multilabel classification problems, which is particularly true in the problem of predicting drug resistance for HIV-1 because different mutations have different impact on resistance. Therefore, to create an effective multilabel classifier to predict HIV-1 resistances, it is critical to clarify the labeling ambiguity on data samples that belong to multiple classes and learn an appropriate scaling factor when we train the classifiers for different classes (Wang et al., 2010b).
In this study, we propose a novel Robust Sample Specific Distance (RSSD) for multilabel data to predict HIV-1 drug resistance, which, as illustrated in Figure 1, is able to explicitly rank the relevance of a training sample with respect to a specific class and characterize the second-order data-dependent statistics of all the classes by class-wise distance metrics. In this study, we note that the proposed RSSD in this article is an application of the instance specific distance (ISD; Wang et al., 2011a–c, 2012, 2016) in single-instance (multilabel) classifications to solve the problem of predicting HIV-1 drug resistance, which was originally proposed in our previous works to solve multi-instance learning problems. We refer the interested readers to Wang et al. (2011a–c, 2012, 2016) for the definition of single-instance learning problems and that of multi-instance learning problems. To learn the sample relevances and the class-specific distance metrics, we formulate a learning objective that simultaneously maximizes and minimizes the summations of the ℓ1-norm distances. To solve the optimization problem of our objective, using the same method in our recent works (Han et al., 2018; Liu et al., 2018), we derive an efficient iterative algorithm with theoretically guaranteed convergence, which, different from our previous works (Wang et al., 2012, 2014), is a nongreedy algorithm such that it has a better chance to find the optima of the proposed objective. We have applied our new method to predict the HIV-1 drug resistance on a public benchmark data set, and the experimental results have shown that our new RSSD method outperforms other state-of-the-art competing methods.
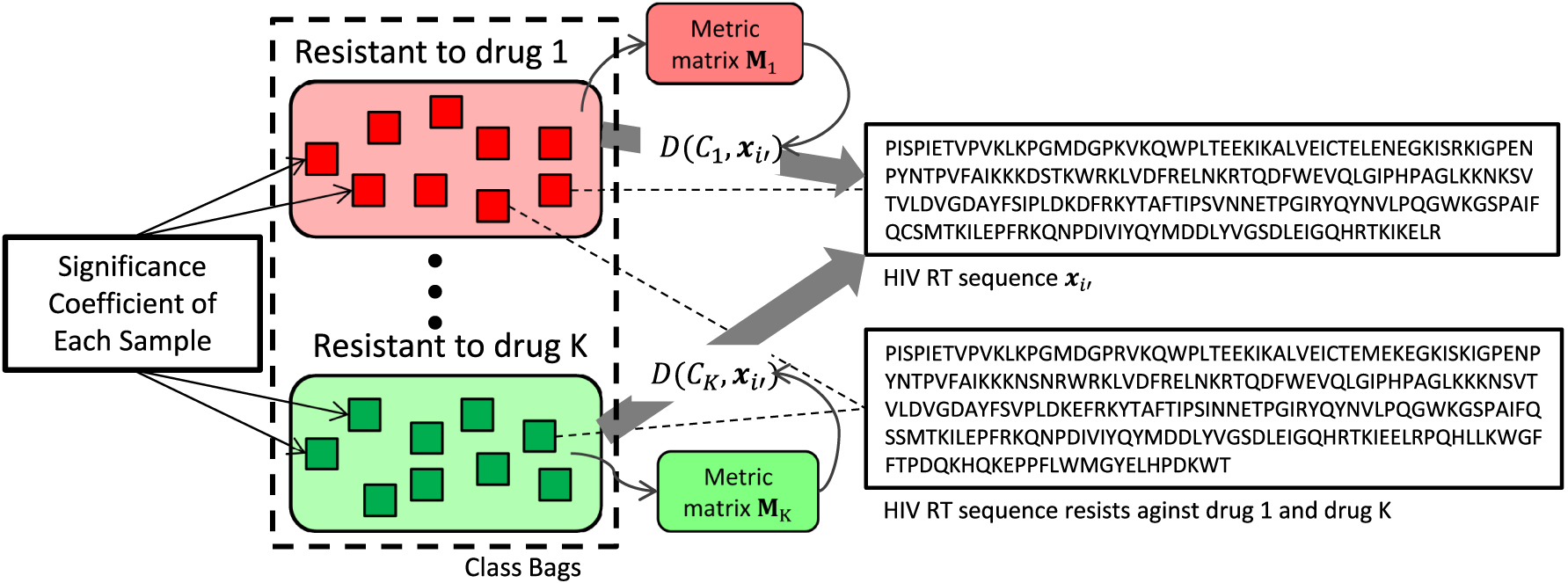
The illustration of the proposed RSSD method. The small squares in the same color represent the data samples (RT sequences) that belong to one same class (e.g., resistance to a specific nucleoside analog). Two HIV RT sequences are listed in the right panel, which correspond to the data samples shown by the small squares connected by the dash lines. The top sequence in the right column only resists against drug 1, while the bottom sequence resists against both drug 1 and drug K, that is, it is a multilabel data sample. Ideally, the learned SCs for each data sample should be different with respect to different classes. For example, the bottom RT sequence is associated with si1 for class 1 and siK for class K, which could be different depending on how the resistances evolved. RSSD, Robust Sample Specific Distance; RT, reverse transcriptase.
Notations and problem formalization
Throughout this article, we write matrices as bold uppercase letters and vectors as bold lowercase letters. The ℓ1-norm of a vector
In a multilabel classification problem, we are given a data set with n samples (n RT sequences)
The class-to-sample distance
To learn the distance from a class to a data sample, we first represent each class as a bag consisting of all samples that belong to this class, that is,
We first define the elementary distance from a data sample
We then compute the class-to-sample (C2S) distance from Ck to
Because the C2S distance in Equation (2) does not take into account the resistance strength against a certain nucleoside analog, we further develop it by weighting the samples in a class bag by their relevance to this class.
Due to the ambiguous associations between the samples and the labels under the multilabel classification setting (Wang et al., 2010a,b), some samples in a class may characterize that particular class more strongly than the others from the statistical point of view. For example, Riemenschneider et al. (2016), where some viral RT sequences may develop a stronger drug resistance, while other viral RT sequences may be less resistant to a drug but may still be considered to be resistant. To develop an effective predictive model for HIV-1 drug resistance development, we need to capture these resistance differences. To be more specific, we should assign less weight to less resistant RT sequences when we determine whether or not to apply the “resistant” label to a query viral RT sequence.
Because we assume that counter-resistance against a target nucleoside analog does not exist, we define sik ≥ 0 as a non-negative constant that assesses relative importance of
Because sik reflects the relative importance of a sample
The RSSD defined in Equation (3) is simply a weighted Euclidean distance that does not take into account the information conveyed by the input data other than the first-order statistics. Similar to many other statistical models in machine learning, using the Mahalanobis distances with appropriate distance metrics is recommended to capture the second-order statistics of the input data. Instead of learning one single global distance metric for all the classes as in many existing statistical studies, we propose to learn K different class-specific distance metrics
Because the class-specific distance metric
A critical problem of
which we call the proposed RSSD.
To use RSSD defined in Equation (6), we need to learn two sets of parameters sik and
Learning the RSSDs by solving Equation (7) and classifying query viral RT sequences using the adaptive decision boundary method (Wang et al., 2009, 2013a), our proposed RSSD method can be used for multilabel classification.
Our new objective in Equation (7) maximizes the ratio of the summations of a number of ℓ1-norm distances, which is obviously not smooth thereby making it difficult to solve in general. To solve this challenging optimization problem, we first turn to solve the following generalized objective:
where Ω is the feasible domain. Next, we propose a simple, yet efficient, iterative framework in Algorithm 1 to solve the objective in Equation (8). The convergence of Algorithm 1 is rigorously guaranteed by Theorem 1.
Proof. In Algorithm 1, from step 3 we have h(vt) − λtm(vt) > 0. Because
Suppose that for the k-th iteration, there exists a ct such that h(vt) − λtm(vt) = ct > 0. We have:
by which we can derive:
From Equation (10), we can derive:
Suppose that there exist a positive constant C such that
Thus, we have:
which means that:
which indicates that:
by which and Equation (9), we have:
which indicates that Algorithm 1 converges to a local optimum and completes the proof of the second statement of Theorem 1.
According to step 3 in Algorithm 1, we can easily write the corresponding inequality of our objective in Equation (7) as:
where λt is computed by
and
In Equation (18),
Now we need to solve the problem in Equation (17), for which we first introduce the following two lemmas:
Motivated by Lemmas 1 and 2, we construct the following objective:
where
Here
and
Then, using the definition of
where the equality holds if and only if
Proof. First, according to Lemma 1 we can compute:
Then, according to Lemma 2 we have:
which indicates that:
Combining Equations (28) and (30), we can derive:
According to Lemmas 1 and 2, it is easy to verify that equality holds in Equations (28) and (30) if and only if
Now we continue to solve our objective. Let
Lemma 1 and Equation (32) indicate that the solution of the objective function in Equation (17) can be transformed to solve the objective function
Finally, based on Algorithm 2, we can derive a simple yet efficient iterative algorithm as summarized in Algorithm 3 to solve our objective in Equation (7) when sik is fixed. In addition, Theorem 3 indicates that our proposed Algorithm 3 monotonically increases the objective function value in each iteration. Theorem 4 indicates that the objective function is upper bounded, which, together with Theorem 3, indicates that Algorithm 3 converges to a local optimum.
Proof. Since
from which we can easily derive:
Now by substituting Equation (18) into Equation (34), we have
which completes the proof of Theorem 3.
Proof. First, using Cauchy–Schwarz inequality we have the following for the numerator of our objective in Equation (7):
Obviously, given an input data set,
Second, it can be verified that
where λi(i = 1, …, r), ordered by λ1 ≤ ⋯ ≤ λr, are the eigenvalues of
The two bounds in Equations (37) and (38) together indicate that our objective in Equation (7) is upper bounded.
When
Defining that
Again, to solve Equation (40), to step 3 in Algorithm 1, we solve the following optimization problem:
where λ is computed as Equation (18) in the t-th iteration.
Define that
The problem in Equation (42) can be decoupled to solve the following subproblems separately for each
which is a convex linear programming problem (Wright and Nocedal, 1999) and can be solved efficiently by many off-the-shelf solution algorithms (Wright and Nocedal, 1999). By inserting the solution to Equation (43) after step 3 of Algorithm 3, we can finally solve our objective in Equation (7), which is equivalent to performing alternative optimization. Therefore, the algorithm is guaranteed to converge to a local optimum.
We evaluate the proposed RSSD method using a publicly available HIV drug resistance database (Rhee et al., 2006), which contains HIV-1 RT sequences with associated resistance factors measured by IC50 ratios. We analyze the drug resistance of these RT sequences against five nucleoside analogs: Lamivudine (3TC), Abacavir (ABC), Zidovudine (AZT), Stavudine (d4T), and Didanosine (ddI). Following Heider et al. (2010), although the Tenofovir nucleoside analog is included in this database, it is not used in our study, because the number of the RT sequences resistant to this nucleoside analog is very small. As a result, we end up with 623 RT sequences for our experiments.
Drug resistance of a particular HIV strain is measured by the IC50 ratio, which is defined as the concentration of a specific drug inhibiting 50% of viral replication compared with cell culture experiments without the drug:
We label the viral RT sequences as “resistant” using the same drug-specific IC50 ratio cutoff thresholds as in Heider et al. (2013), which are set to 3.0 for 3TC and AZT, 2.0 for ABC, and 1.5 for ddI and d4T. We use hydrophobicity characteristics (Kyte and Doolittle, 1982) to represent the RT sequences, which have demonstrated good prediction performance in many protein classification studies (Heider et al., 2010). For each RT sequence, we extract a hydrophobicity vector, which is obtained from the amino acid sequence and smoothed within a window. The length of the original hydrophobicity vectors may be different due to the different lengths of the RT sequences. In this study, following Heider et al. (2013) we set a fixed window size of 11 and interpolated all hydrophobicity vectors to length 230 using the spline interpolation method (Kyte and Doolittle, 1982).
In Section 3, we have theoretically proved the convergence of the derived solution algorithm. Now we study the convergence of our new algorithm from the empirical perspective. We apply our new method on the HIV-1 drug resistance data set and plot the objective value after each iteration in Figure 2. This plot clearly shows that our solution algorithm converges very fast and confirms the correctness of our new method.
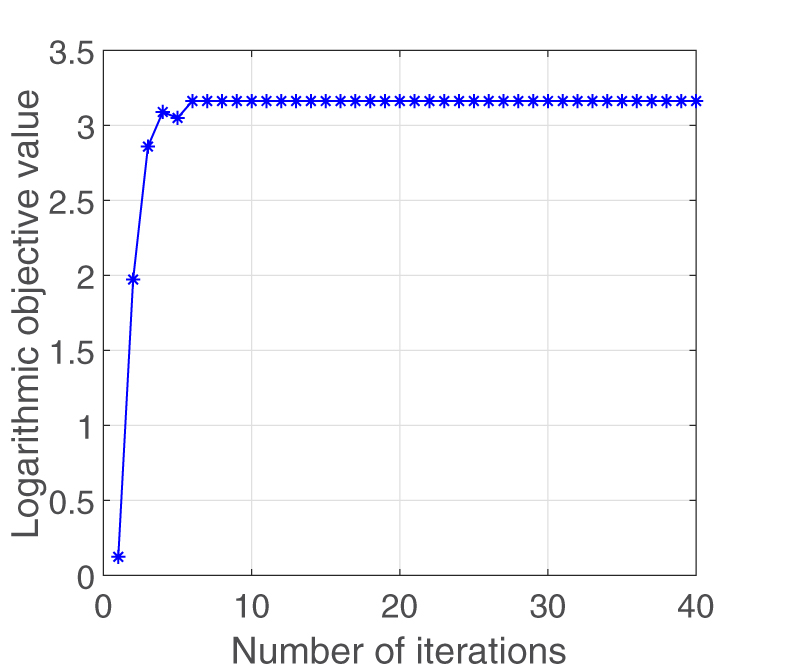
The convergence of our solution algorithm.
Predicting drug resistance for HIV-1 RT sequences is a multilabel classification problem. Therefore, we evaluate the proposed method by two broadly used multilabel performance metrics (Lewis et al., 2004): Hamming loss and average precision. The Hamming loss is computed over all instances over all classes. The average precision is calculated for both the micro and macro averages. In multilabel classification, the macro average is computed as the average of the precision values over all the classes; thus, it attributes equal weights to every individual class. In contrast, the micro average is obtained from the summation of contingency matrices for all binary classifiers, thus it gives equal weight to all classifiers and emphasizes the accuracy of categories with more positive samples.
The proposed RSSD has only one parameter: the dimensionality r of
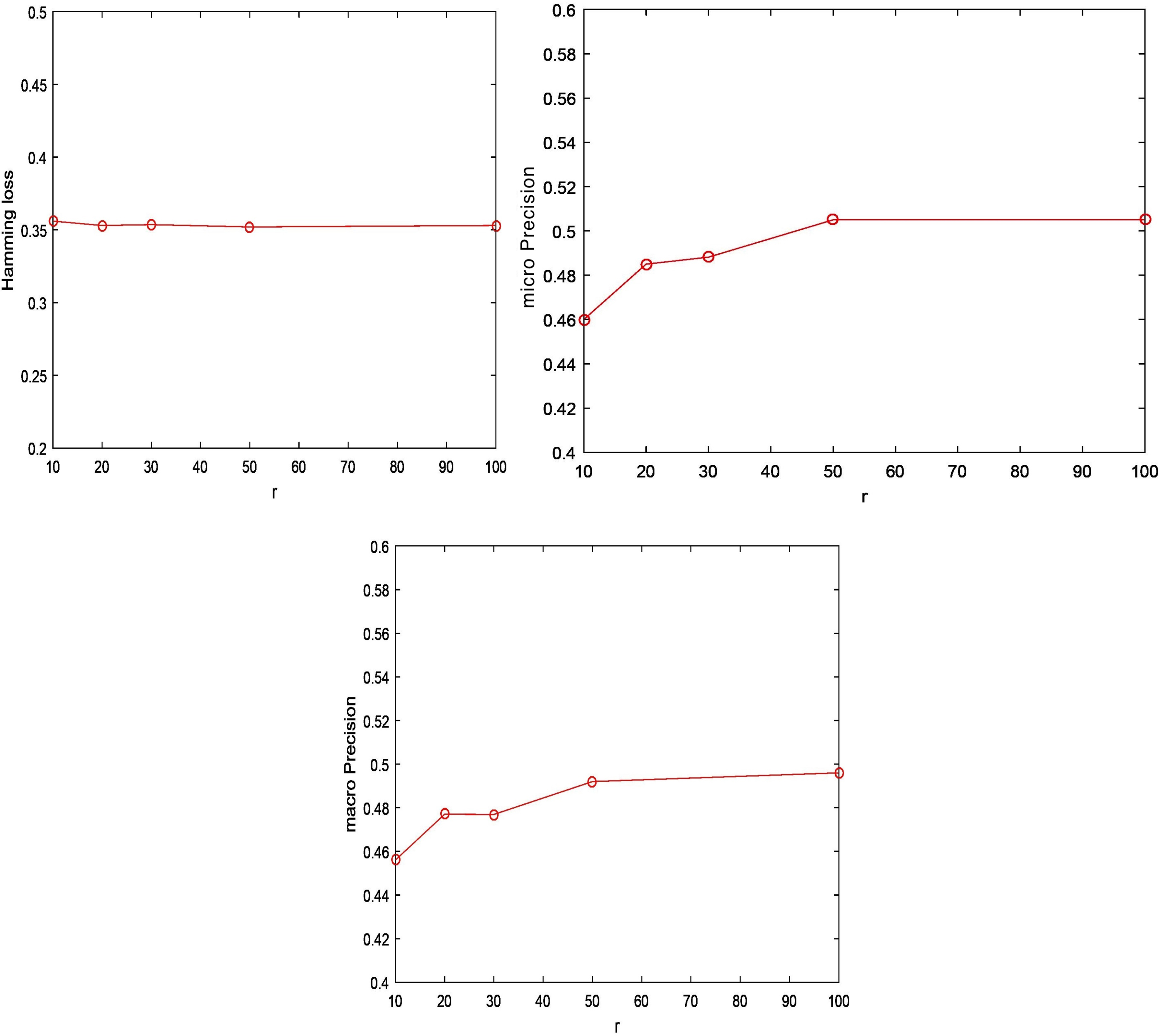
Multilabel classification performance of the proposed method on the HIV-1 drug resistance data with respect to r (the dimensionality of
We use a standard fivefold cross-validation to evaluate the predictive capability of the proposed RSSD method. We implement two versions of our proposed method, one version that defines
We also compare the proposed method against two multilabel classification methods designed to study drug resistance in HIV-1: the classifier chain (CC) method and its ensemble version (Read et al., 2011; Heider et al., 2013; denoted as the ensemble of classifier chain [ECC] method). Finally, we also compare our method to two recent multi-instance classification methods: the MTL method (Yuan et al., 2016) designed to study general drug resistance study and the deep multi-instance multilabel (MIML) method (Feng and Zhou, 2017) designed to study general multi-instance data. The Green's Function method and the SMSE methods are implemented following their original articles in Wang et al. (2009) and Chen et al. (2008), respectively, where the parameters are set to the suggested values. The CC method is implemented with logistic regression, where the chaining order for the CC method is 3TC → ABC → AZT → d4T → ddI as suggested in Heider et al. (2013). Following Heider et al. (2013) and Riemenschneider et al. (2016), we implement the ECC method using both random forests and logistic regression as base classifiers, which are denoted as “ECC-RF” and “ECC-LR,” respectively. The MTL method and the deep MIML method are implemented using the code published by the respective authors. The overall resistance prediction performances of the compared methods are reported in Table 1.
Performance of the Compared Methods by Standard Fivefold Cross Validations
Performance of the Compared Methods by Standard Fivefold Cross Validations
Where “↓” means that smaller is better, and “↑” means that bigger is better.
CC, classifier chain; ECC, ensemble of classifier chain; MIML, multi-instance multilabel; MTL, multitask learning; SMSE, Sylvester equation.
The comparison results in Table 1 show that the ℓ1-norm version of the proposed method consistently outperforms all competing methods in terms of all the three performance metrics, sometime very significantly. The squared ℓ2-norm version of our new method is, as expected, not as effective as its counterpart using the ℓ1-norm distance, but it still provides adequate performance compared to the other methods in Table 1.
We explore the learned distances by our method between RT sequence pairs and compared them with the Euclidean distances for the same RT sequence pairs. The distance between two RT sequences by our method is defined as the sum of the two learned RSSDs: for the k-th class, the pairwise distance between sequence
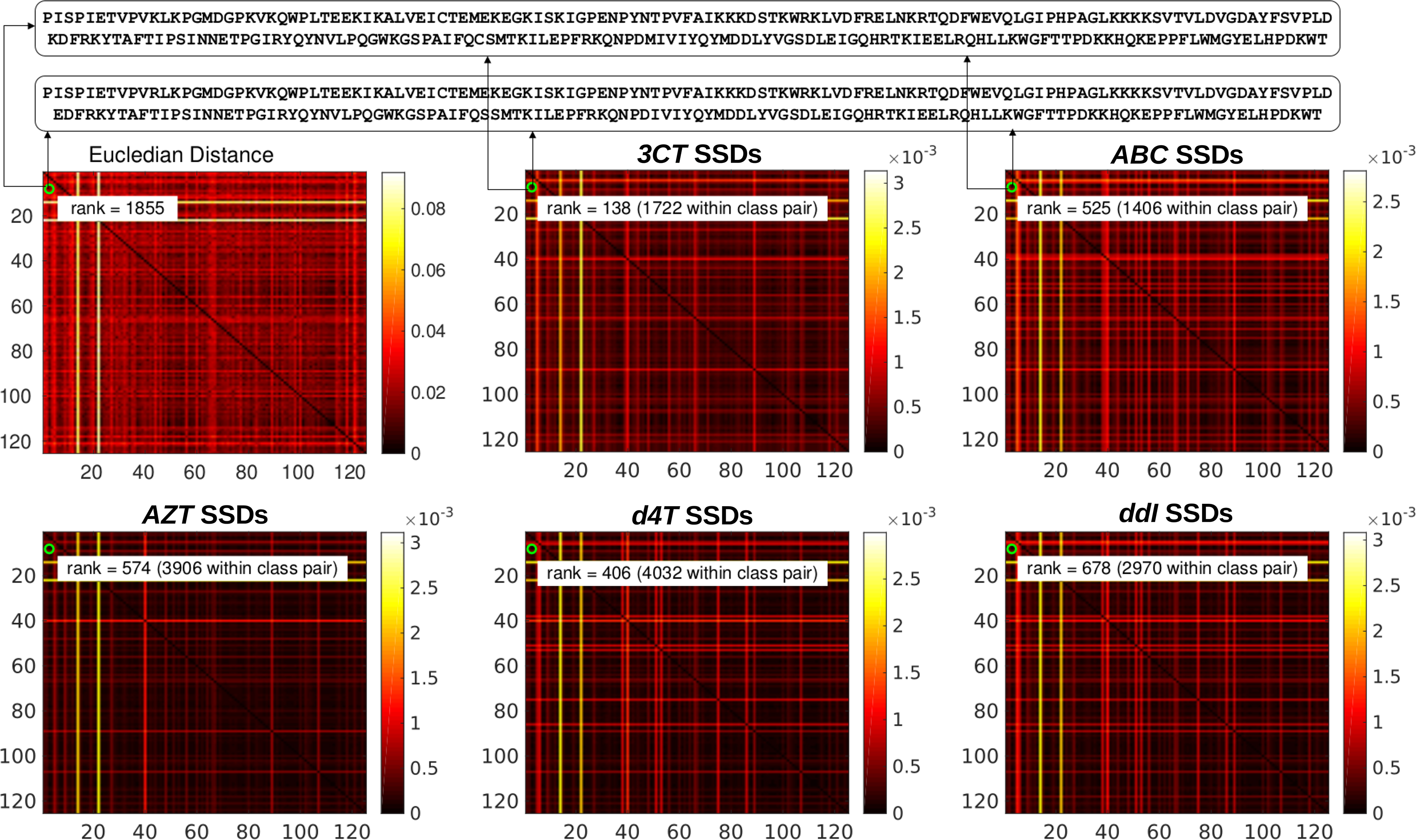
Exploration of the learned sample-to-sample distance between RT sequence pairs for each class. Top panel: The two RT sequences (with known drug resistance) we are comparing; Top Left Heatmap: the Euclidean distances between RT sequence pairs. Remaining Heatmaps: the learned sample-to-sample distances between RT sequence pairs for each of the five classes. We can see that the sample-to-sample distance between the two RT sequences in the top panel for 3CT nucleoside analog is ranked as the 138-th smallest pairwise distance among all 1722 RT sequence pairs. Compared to the Euclidean distance, which is ranked as 1855-th smallest distance, the pairwise distance computed by the projection and significance coefficients learned for this class is more clinically meaningful. 3TC, Lamivudine; ABC, Abacavir; AZT, Zidovudine; d4T, Stavudine; ddI, Didanosine; SSD, sample specific distance.
To demonstrate the effectiveness of the proposed method, we study the distances between two example RT sequences, which are listed at the top of Figure 4. These two RT sequences are known to be resistant to all five nucleoside analogs. As a result, the pairwise distance between these two RT sequences is expected to be small. However, as can be seen in top left panel of Figure 4, the Euclidean distance between these two RT sequences is ranked at the 1855-th smallest distance among all pairwise Euclidean distances, which is not in accordance with the clinical evidences. In contrast, we can see that the pairwise distances between these RT sequences computed by our learned RSSDs for the five classes are small, which are at the 138-th smallest distance for 3TC, the 525-th smallest distance for ABC, the 574-th smallest distance for AZT, the 406-th smallest distance for d4T, and 678-th smallest distance for ddI, respectively. This observation clearly demonstrates that the learned distances by our new methods can better capture the relationships between data samples in terms of class membership.
From the subspace learning perspective, the ISD in Equation (6) can be written as
which is the learned C2S distance in the projected lower r-dimensional subspace, where the projection is implemented by
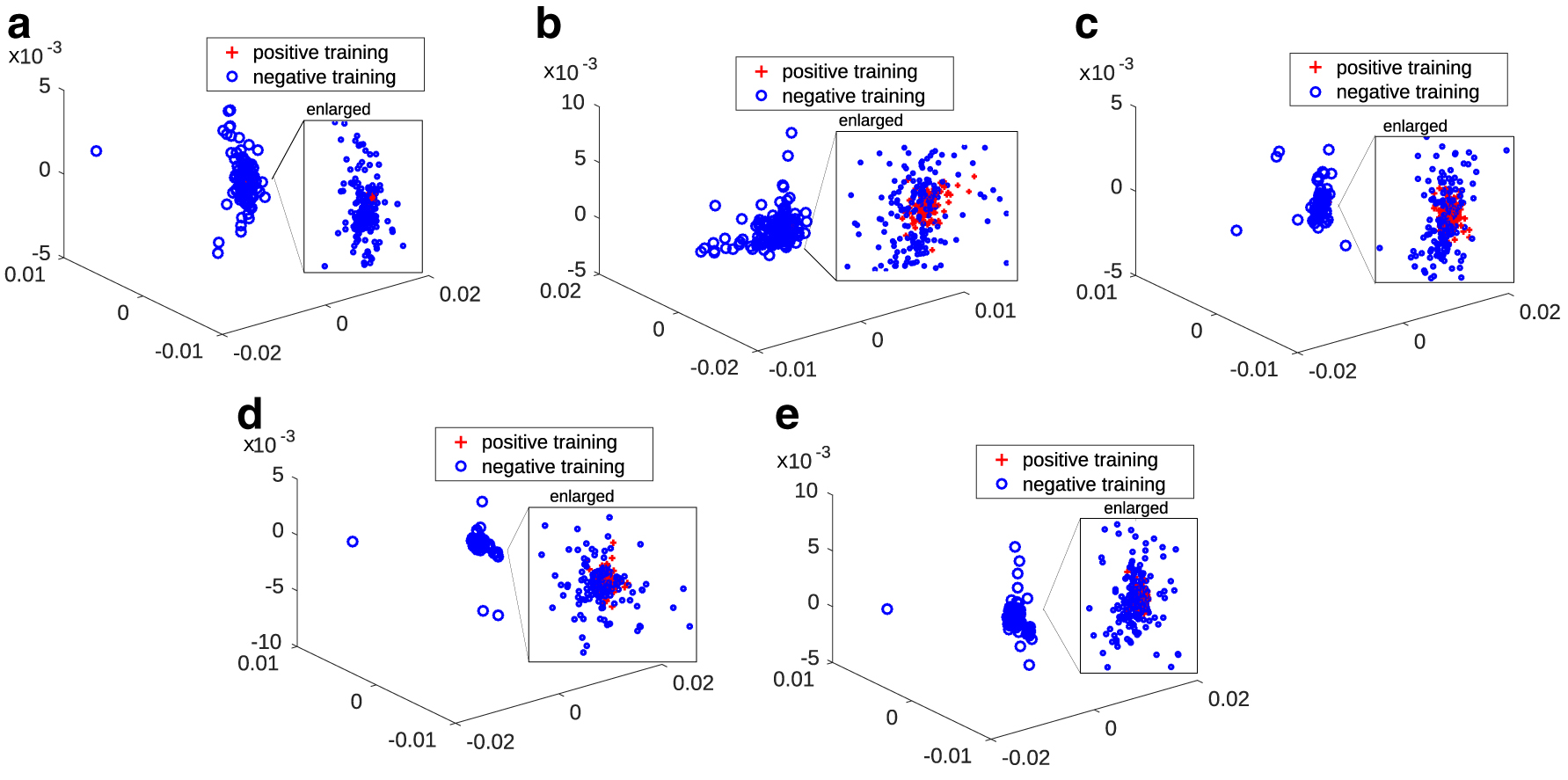
Projected data samples through the learned projection matrix
From Figure 5 we can see that the proposed method works very well in terms of maximizing data separability. In each of the five projected subspaces, data samples belonging to any one of target classes are close to one another, while those not belonging to the same class are far away from each other. This observation concretely suggests that the learned projected matrices
To further evaluate the discriminative ability of the proposed method, we plot the top 10 shortest RSSDs among test data for every class in Figure 6. From the results we can see that, shown by red crosses in each figure, most data samples with the smallest RSSDs indeed belong to the target class. This observation once again suggests that the proposed RSSDs with optimized distance metrics and instance-specific SCs are good criteria for drug resistance prediction.
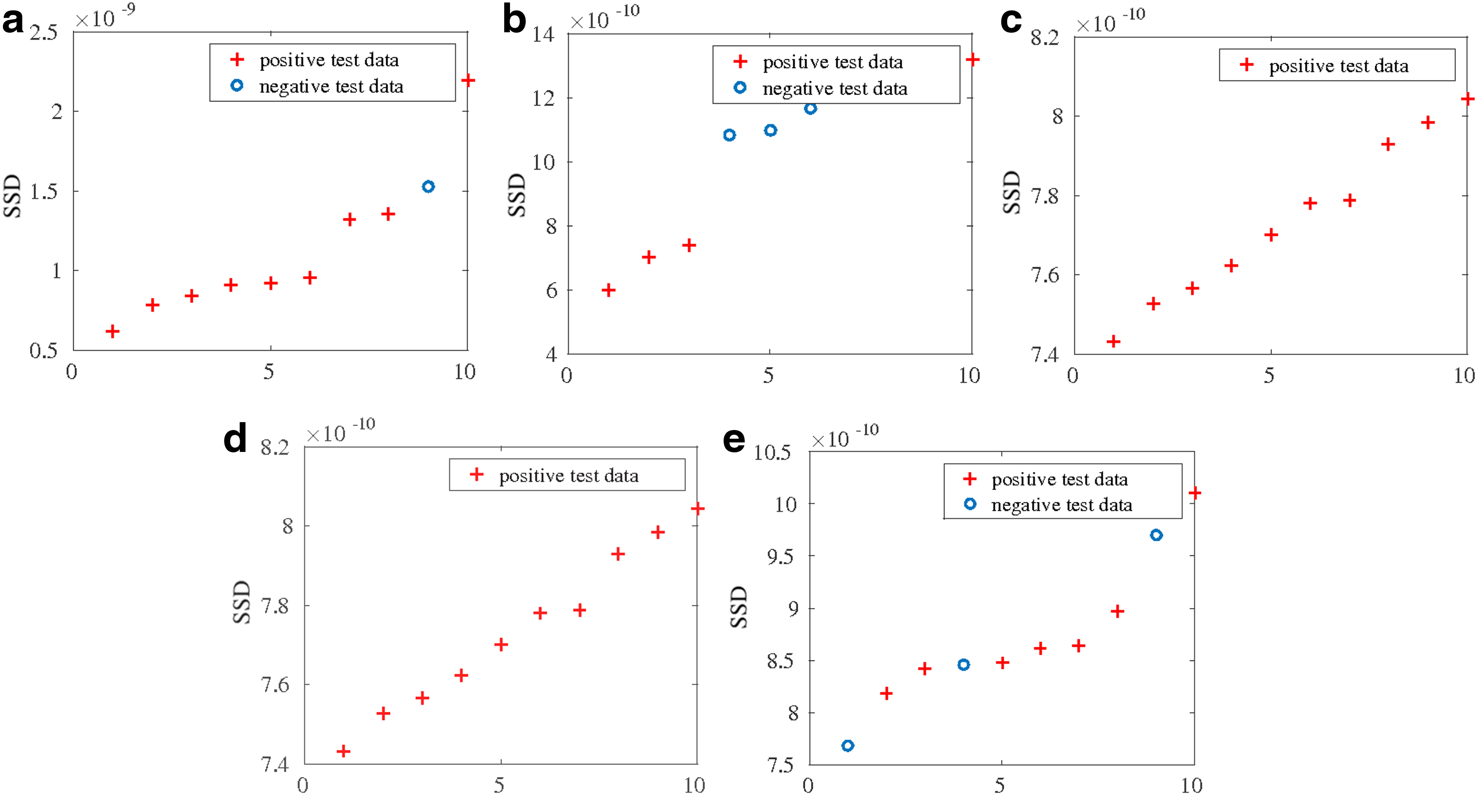
True labels of top 10 data with smallest RSSDs for each of the five classes.
In this study, we proposed a novel RSSD method for multilabel classification. To learn the parameters of the proposed RSSDs, we formulated a learning objective that maximizes the ratio of the summations of a number of ℓ1-norm distances, which is difficult to solve in general. To solve this problem we derived a new efficient iterative algorithm with rigorously proved convergence. The promising experimental results have demonstrated the effectiveness of our new method for identifying HIV-1 drug resistances.
Footnotes
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This work was partially supported by the National Science Foundation under the grants of IIS-1652943 and IIS-1849359.