
Abstract
Cardiovascular and cerebrovascular diseases, which mainly consist of atherosclerosis (AS), are major causes of death. A great deal of research has been carried out to clarify the molecular mechanisms of AS. However, the etiology of AS remains poorly understood. To screen the potential genes of AS occurrence and development, GSE43292 and GSE57691 were obtained from the Gene Expression Omnibus (GEO) database in this study for bioinformatic analysis. First, GEO2R was used to identify differentially expressed genes (DEGs) and the functional annotation of DEGs was performed by gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. The Search Tool for the Retrieval of Interacting Genes (STRING) tool was used to construct the protein–protein interaction network and the most important modules and core genes were mined. The results show that a total of 211 DEGs are identified. The functional changes of DEGs are mainly associated with the cellular process, catalytic activity, and protein binding. Eighteen genes were identified as core genes. Bioinformatic analysis showed that the core genes are mainly enriched in numerous processes related to actin. In conclusion, the DEGs and hub genes identified in this study may help us understand the potential etiology of the occurrence and development of AS.
1. Introduction
Atherosclerosis (AS)
At present, the research on AS is mainly about cardiovascular and cerebrovascular diseases. In addition to the classical lipid deposition theory and the classical inflammatory response involving monocytes/macrophages, neutrophils, and T cells, allergic inflammatory effector cells have also been found to be involved in the formation of AS (Niccoli et al., 2018). At present, it is emphasized that the preventive effect can be achieved by controlling some risk factors of AS, such as low-density lipoprotein and cholesterol, smoking, and blood pressure. After the formation of AS, antiplatelet aggregation and blood lipid-lowering drugs are applied. The local vascular stent or bypass measures are also used to improve tissue and organ ischemia caused by AS. However, the therapeutic effect of the above treatment measures in the treatment of AS is still not found to be satisfactory. The morbidity and mortality of AS are still high, which present a serious threat to human health (Kelly et al., 2018). There has been no milestone progression such as for aspirin in the treatment of AS. The perfect therapeutic drugs or measures have not been found, which suggests that we need to further study the pathogenic genes or molecules of AS, providing evidence for targeted therapy and early gene therapy for AS.
Bioinformatics is one of the frontier areas of life sciences and natural sciences. It stores, retrieves, and analyzes various complex biological data using the computer, which is a new interdisciplinary between life science and computer science. Bioinformatics focuses on genomics and proteomics. It starts from the nucleic acid and protein sequence, analyzes the biological information on structural function in the sequence, and finds genes and proteins related to diseases. Nowadays, more and more researchers use bioinformatics to find the relationship between diseases and gene sequences, especially molecular mechanisms related to the targeted treatment. By comparing and analyzing the RNA sequences of pancreatic islets in diabetic and nondiabetic patients, Kameswaran et al. (2014) found that microRNAs and DNA methylation may be involved in the pathogenesis of diabetes. Zheng found the PSAT1 gene related to ovarian carcinoma through bioinformatic technology mining from the large public database and verified its correlation through further experiments, suggesting that it serves as a diagnostic and therapeutic target (Zhang et al., 2018). Therefore, bioinformatic analysis can be applied to find target molecules for treatment of the disease.
We screened the diseased blood vessels of patients with AS and compared them with normal human blood vessels. Gene ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis were performed on the corresponding genes and molecules, and then the significant module of the protein-protein interaction (PPI) network was screened. We identified genes that were differentially expressed between the diseased blood vessels of patients with AS and normal human blood vessels and further identified the most relevant genes. These findings will improve our understanding of the underlying mechanisms of AS and provide the basis for finding new targeted therapies.
2. Materials and Methods
2.1. Access to public data
GEO (www.ncbi.nlm.nih.gov/geo) (Wang et al., 2016) is an open-source, high-throughput genomic database that includes microarrays, gene expression data, and chips. Two expression profile data sets (GSE43292 and GSE57691) were downloaded from the GEO database. The annotation platform for GSE43292 is the GPL6244 platform, [HuGene-1_0-st] Affymetrix Human Gene 1.0 ST Array [transcript (gene) version]. The GSE43292 data set comprises 32 atherosclerotic vascular tissues and 32 nonatherosclerotic vascular tissues. The annotation platform for GSE57691 is the GPL10558 platform, Illumina HumanHT-12 V4.0 expression beadchip. The GSE57691 date set comprises 10 atherosclerotic vascular tissues and 9 nonatherosclerotic vascular tissues. All probe numbers are converted to gene symbols based on the annotation information in the platform.
2.2. Screening of differentially expressed genes through GEO2R
GEO2R (www.ncbi.nlm.nih.gov/geo/geo2r) (Barrett et al., 2013) is a system for online analysis of data in GEO. This tool runs using the R language. It is accurate to say that it uses two R packages: GEOquery and limma. The former is used for data reading and the latter is used for calculation. The best thing about GEO2R is that it is an online tool that is easy and efficient to operate. GEO2R can perform a command to compare gene expression profiles between groups to identify differentially expressed genes (DEGs) between atherosclerotic and nonatherosclerotic groups. In general, when the probe set has a corresponding gene symbol, the probe is considered valuable and will be retained. Statistically significant measures are adj. p-value ≤0.01 and fold change ≥1.5.
2.3. Functional annotation of DEGs through GO and KEGG analysis
Database for Annotation Visualization and Integrated Discovery (DAVID) (https://david.ncifcrf.gov/home.jsp; version 6.8) is a bioinformatic database that integrates biological data and analytical tools to provide systematic and comprehensive annotation information on biological functions of large-scale genes, or protein lists to help users extract biological information from them (Huang et al., 2007). KEGG (www.kegg.jp) is one of the most commonly used bioinformatic databases in the world to understand advanced functions and biological systems. At the molecular level, KEGG integrates a large number of utility database resources generated by high-throughput experimental techniques (Kanehisa, 2002). GO is widely used in bioinformatics, which covers three aspects of biology, including cellular components (CCs), molecular function (MF), and biological process (BP) (Ashburner et al., 2000). To analyze the GO and pathway enrichment information of DEGs, the DAVID online tool was executed. A statistically significant measure is p < 0.05.
2.4. Construction and analysis of the protein–protein interaction network
The Search Tool for the Retrieval of Interacting Genes (STRING; http://string-db.org; version 10.5) (Szklarczyk et al., 2015) is a network that can be used to predict and track protein–protein interactions. Intermolecular network analysis can be done by introducing DEGs into the tool. The analysis of interactions between different proteins can provide some new ideas for the study of pathophysiological mechanisms of AS. In this study, the STRING database was used to construct the PPI network with DEGs. The minimum required interaction score is medium confidence >0.4. Cytoscape (version 3.6.1) is an open-source visualization software tool that can be used to visualize the protein–protein interaction network (Su et al., 2014). Based on topological principles, Molecular COmplex DEtection (MCODE) (version 1.5.1), a plug-in for Cytoscape, can mine tightly coupled regions from PPIs. First, Cytoscape software plots the PPI network. Second, MCODE identifies the most important modules in the PPI network graph. The criteria for MCODE analysis are as follows: node score cutoff = 0.2, degree cutoff = 2, max depth = 100, MCODE score >5, and k-score = 2.
2.5. Mining and screening of core genes
A total of 18 genes were identified as core genes with degrees ≥ 18. The core gene coexpression networks in the field of atherosclerotic diseases were analyzed using the open-source online tool Coexpedia (www.coexpedia.org) (Yang et al., 2017). BPs of core genes were analyzed and visualized using the Biological Network Gene Ontology Tool (BiNGO; version: 3.0.3) (Maere et al., 2005). The expression profiles of tropomyosin 2 (TPM2) in the normal organ cells of Homo sapiens were analyzed and displayed using the online database (Raw Data Provider: The FANTOM5 project; www.ebi.ac.uk/gxa/home#search-atlas) (Chen et al., 2002).
2.6. Statistical analysis
Student's t test was used to determine the statistical significance when comparing the two groups. Statistical analysis was carried out using SPSS software, version 21.0 (IBM Corp. Armonk, NY). Values of p < 0.05 were considered statistically significant.
3. Results
3.1. DEGs were identified between nonatherosclerotic and atherosclerotic groups
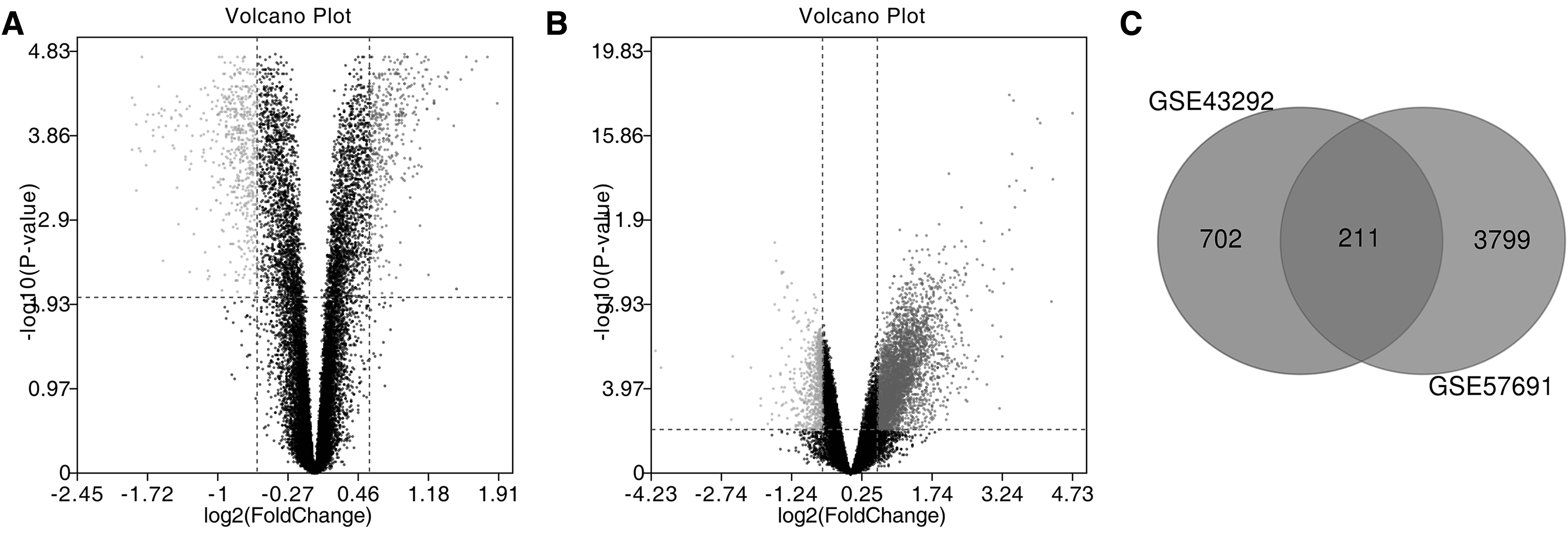
Analysis of the GSE43292 and GSE57691 datasets by GEO2R (volcano plot) showed the differences between control and atherosclerotic tissues (Fig. 1A, B). Then, these results were standardized to distinguish DEGs (913 DEGs were further screened in GSE43292 and 4010 DEGs were further screened in GSE57691). The VENN plot showed that 211 DEGs are included in both data sets (Fig. 1C).

3.2. Functional annotation of DEGs through GO and KEGG analysis
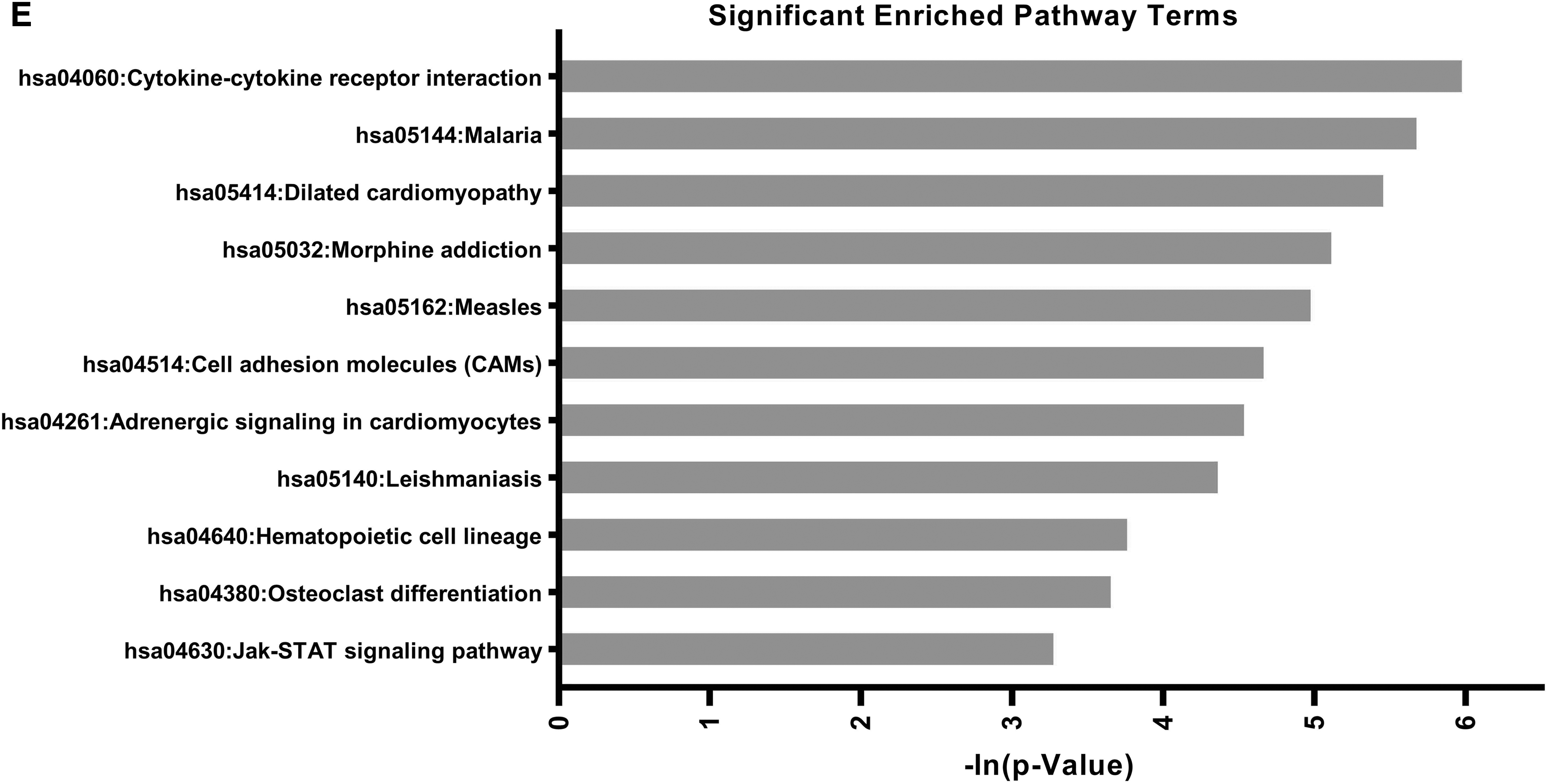
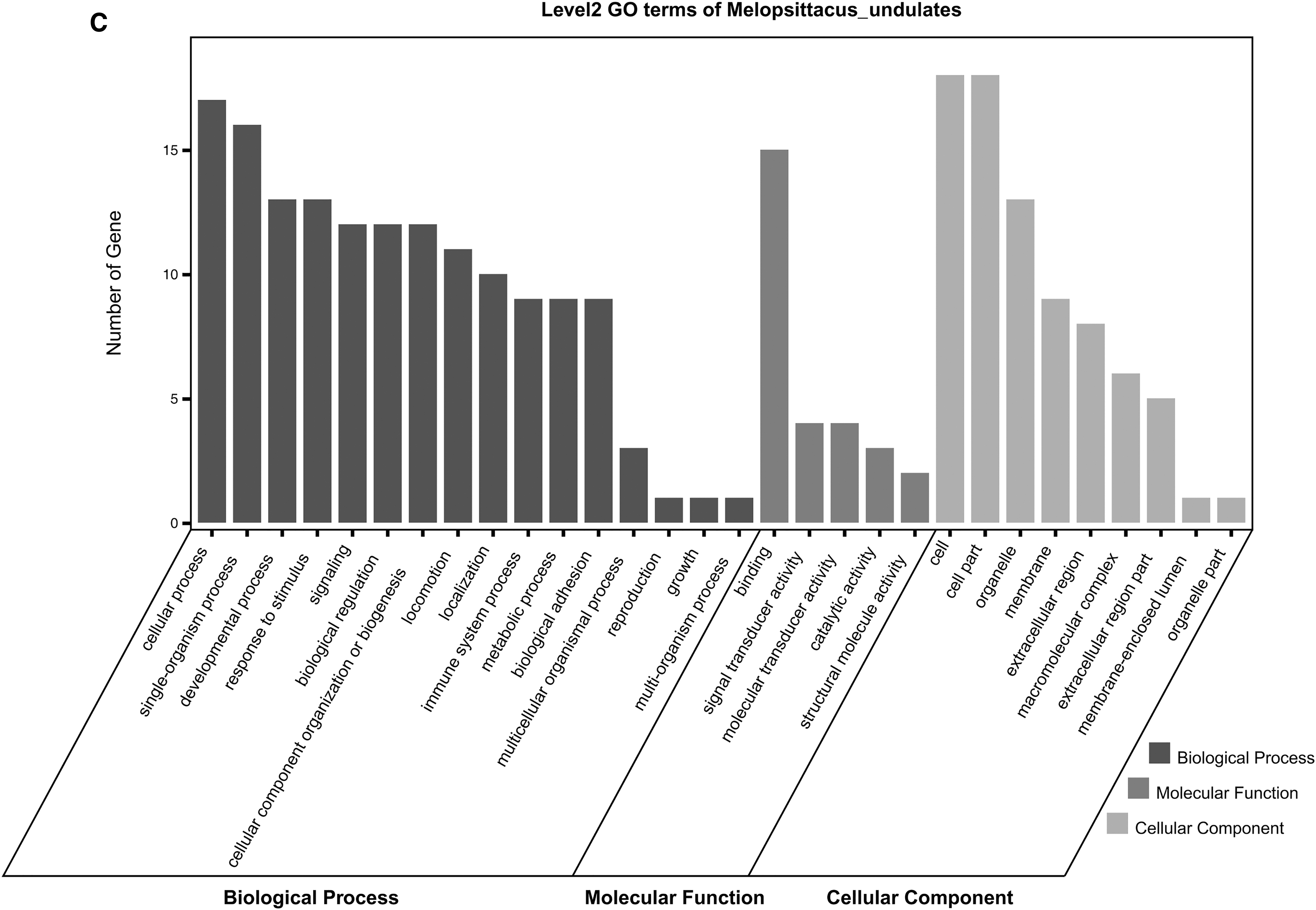
The GO analysis results of changes of BPs showed that DEGs are mainly enriched in the cellular process, CC organization or biogenesis, and multicellular organismal process (Fig. 1D). The changes of MFs showed that DEGs were significantly enriched in binding, catalytic activity, signal transducer activity, transcription factor activity, and protein binding (Fig. 1D). The changes of CCs showed that DEGs were mainly enriched in the cell, cell part, extracellular region, extracellular region part, and extracellular matrix (Fig. 1D). The KEGG pathway analysis showed that all DEGs are mainly concentrated in cytokine–cytokine receptor interaction, malaria, dilated cardiomyopathy, and cell adhesion molecules (Fig. 1E).
3.3. Construction of the PPI network and analysis of significant modules
Construction of the PPI network of DEGs (Fig. 2A) and identification of the most significant modules were done using Cytoscape (Fig. 2B). Functional analysis of the genes involved in this module was performed using DAVID. The results showed that the genes of the module are mainly enriched in the cellular process, binding, cell, and cell part (Fig. 2C). The KEGG pathway analysis showed that the genes in the significant modules are mainly enriched in African trypanosomiasis, cytokine–cytokine receptor interaction, and hematopoietic cell lineage (Fig. 2D).


3.4. Screening and analysis of core genes
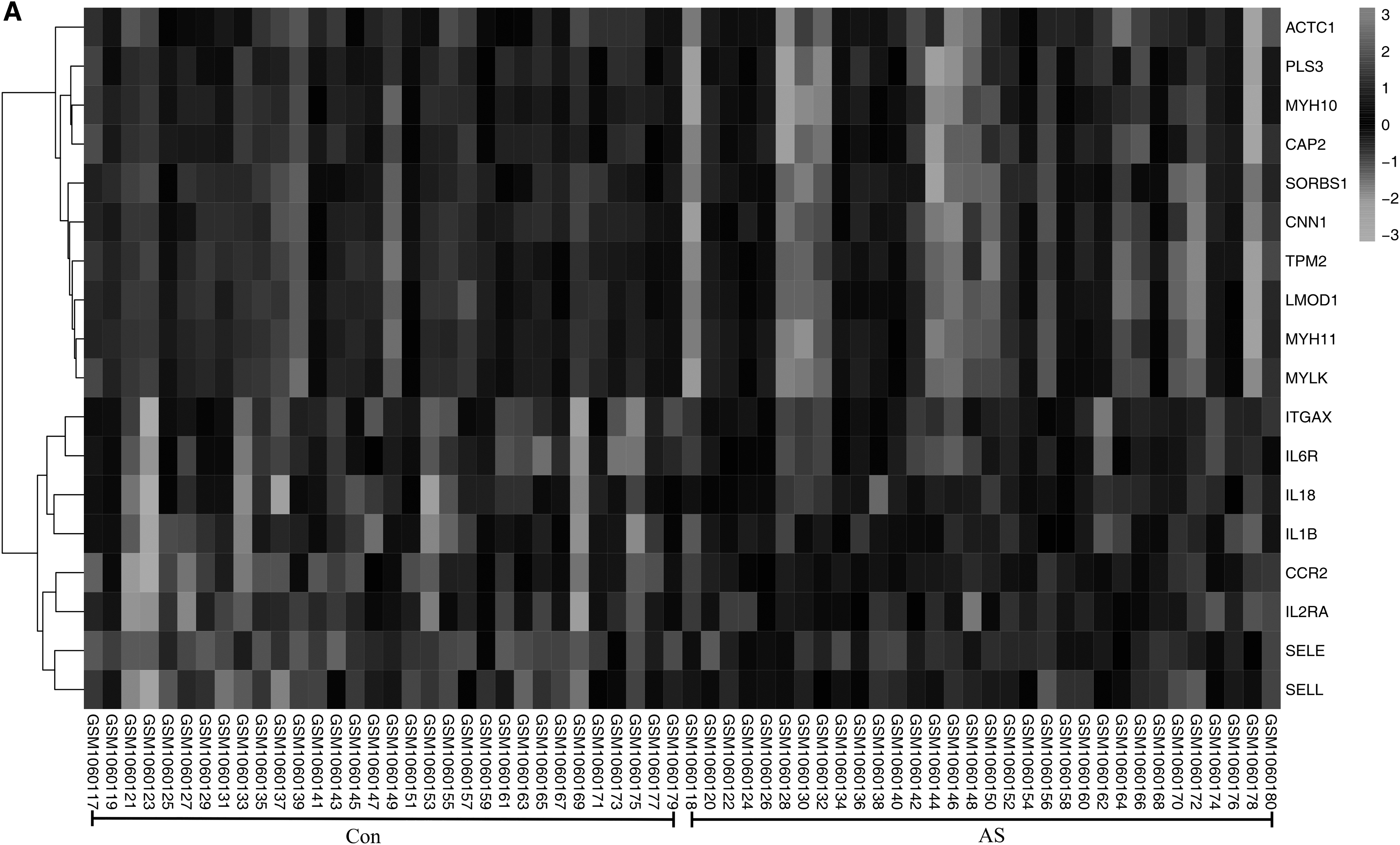
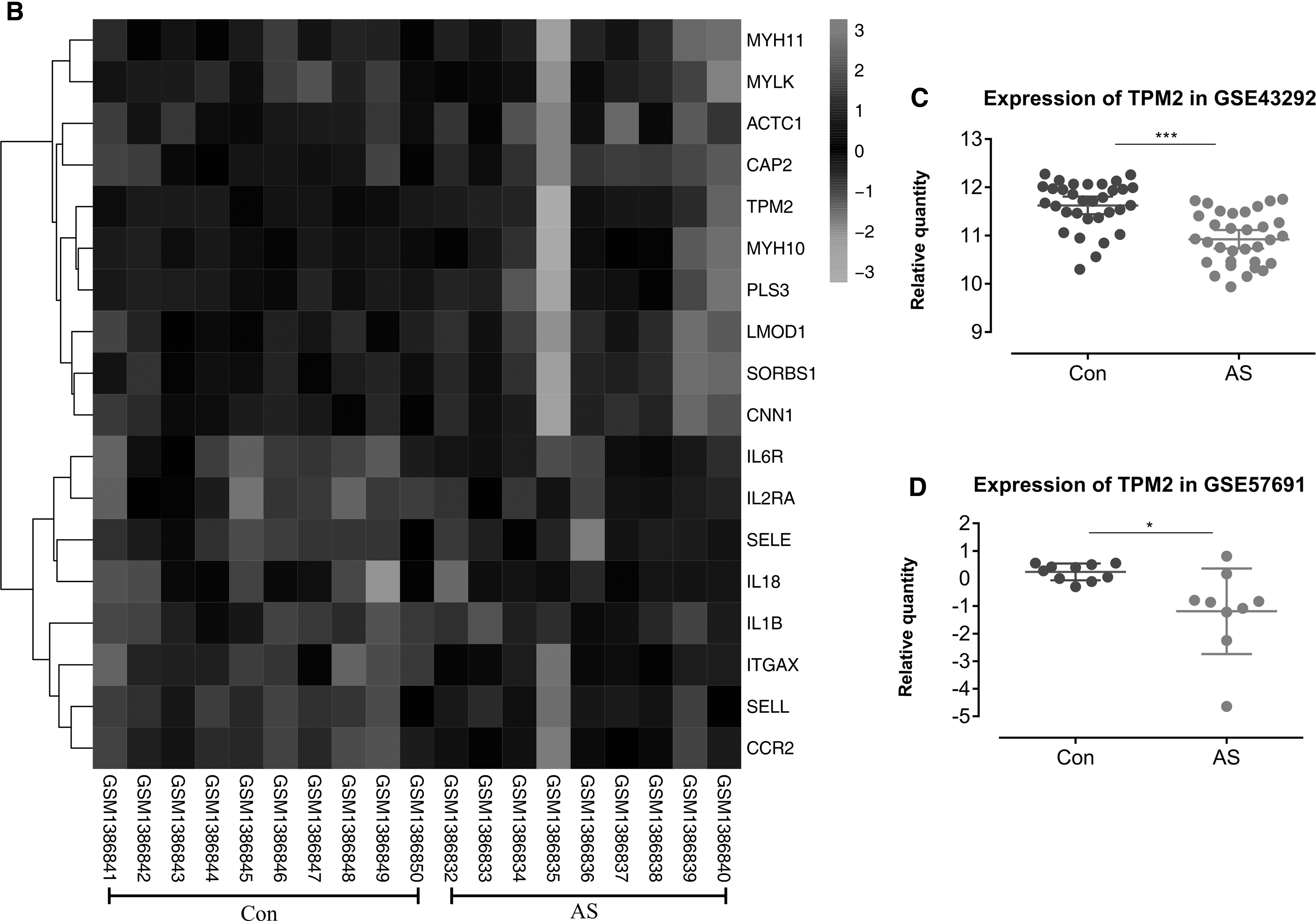
When degree ≥18, a total of 18 genes were identified as core genes. The names, abbreviations, and functions of these core genes are shown in Table 1. The core genes and their coexpression gene networks were analyzed using the Coexpedia online platform (Fig. 3A). The BP analysis of core genes is shown in Figure 3B. The hierarchical clustering results showed that the core genes can distinguish between atherosclerotic and nonatherosclerotic groups (Fig. 4A, B). We found that the expression level of TPM2 in atherosclerotic tissues is higher than that in nonatherosclerotic groups (p < 0.05) (Fig. 4C, D). Furthermore, the expression profile of TPM2 in human tissues was established using the online database (Raw Data Provider: The FANTOM5 project). We found that the expression levels of TPM2 are higher in arteries in the normal population (Fig. 5A). In addition, in the Protein Data Bank (PDB; www.rcsb.org), the structure of TPM2 could be constructed from different visual angles. The 1.8-Å structure of sm-Tmα98 (98-aa) unexpectedly reveals an antiparallel coiled coil, with the two chains staggered by only four amino acids and displaying hydrophobic core interactions similar to those of the parallel dimer. (Fig. 5B).

Interaction network and biological process analysis of hub genes.

Summary of the Functions of 18 Hub Genes
TREGs, regulatory T cells.
4. Discussion
AS can cause various diseases, especially coronary heart disease, with extremely high morbidity and mortality. Cardiovascular and cerebrovascular diseases cause great economic losses to the society and are also common diseases threatening the health of the elderly. At present, the treatment of AS-related diseases is mainly the use of lipid-lowering drugs and anticoagulant drugs. However, the therapeutic effect is limited. The morbidity and mortality due to atherosclerosis-related diseases remain high (Gistera and Hansson, 2017; Kim et al., 2017). The clinical efficacy and safety of other treatment methods such as methotrexate remain to be further studied (Ridker et al., 2019). Therefore, it is of important clinical value to further explore the mechanism of AS and search for target therapeutic molecules.
Bioinformatic technology has been widely used to search for genes and molecules related to the occurrence and development of diseases and is considered to be a promising technology for finding targeted treatments. The technology can be used to find disease-related data from large public databases for analysis and identify the genes or molecules that are most relevant to the disease. Hypertension is similar to AS, with a large number of patients, a long course of disease, and many complications. Cao et al. (2016) found that bioinformatic technology can be applied to screen genes associated with hypertension. Zhu et al. (2019) found and analyzed the molecular miR-140-5p related to pulmonary hypertension through bioinformatic technology from the public database. The future study verified that this molecule mediates the occurrence and development of pulmonary hypertension-related diseases by affecting the function of pulmonary artery smooth muscle cells. Through analysis, we found that TPM2, actin alpha cardiac muscle 1 (ACTC1), and Plastin 3 (PLS3) expression levels are low in the diseased vascular tissues of patients with AS, while C-C motif chemokine receptor 2 (CCR2) expression is high in the diseased vascular tissues of patients with AS.
TPM2, tropomyosin 2, an important molecule in the TPM family, is a rod-shaped protein with a dimer alpha helix structure that is widely present in various eukaryotes and is mainly involved in myofilament gliding and muscle excitatory contraction. TPM plays an important role not only in myofilament gliding but also in the dynamic cytoskeleton system. Moreover, the distribution of TPM isomers in specific cells involved in cellular functions has attracted more and more attention (Kee and Hardeman, 2008). Clarkson et al. (2004) found that TPM2, actin, and troponin are mainly associated with the pathogenesis of congenital myopathy. Bailey et al. (2017) found that TPM is the molecular target leading to high-throughout screening (HTS). Bartelt et al. (2017) found that HTS has the effect of preventing the transformation of arterial into atherosclerotic arteries. On the one hand, HTS can reduce residual lipoproteins before AS, and on the other hand, HTS can also improve high-density lipoprotein (HDL) remodeling and promote cholesterol clearance. Moreover, in terms of lipid deposition, HTS can also increase the apoptosis of macrophages and reduce formation of foam cells (Bartelt et al., 2017). Endothelial function remains an important aspect of formation of AS. Abnormal expression of myosin microfilaments can damage endothelial cells and provide attachment points for lipids and calcium. It also stimulates fibroblast proliferation and exacerbates fibrosis.
The role of the TPM family is to regulate and stabilize myosin microfilaments. Moreover, the TPM family also stabilizes the function of endothelial cells. The effect is determined by the balance of the isomers in cells (Kubo et al., 2013), suggesting that the low expression of TPM2 in the diseased arteries may affect the balance of the isomers in arterial cells and disrupt the function of endothelial cells. Smooth muscle dysfunction is still a factor causing AS. TPM can maintain the stability of smooth muscle actin and affect the vasoconstriction function indirectly. Therefore, the lack of TPM may limit vasoconstriction (Prunotto et al., 2015). Excessive vasodilation can cause the rupture and degeneration of the media layer of arterial walls, which will induce the changes of AS (Ribalet et al., 2005). Different from rodent Tpm1α/2β, which is involved in endothelial cell interstitial fibrosis, we found that TPM2 plays a protective role in the progression of AS through verification of TPM2 molecules using bioinformatic analysis and animal level.
We speculate that on the one hand, TPM2 is highly sensitive in human arteries, and on the other hand, TPM2 may be an important factor in maintaining the stability of the TPM family of isomers in the arteries. In conclusion, we found that TPM2 expression is low in patients with AS. We hypothesized that TPM2 may reduce the formation of intravascular foam cells, improve HDL remodeling, and optimize HDL function through HTS. In addition, TPM2 can stabilize the structure and function of vascular endothelial cells and control the contractile function of smooth muscle cells. These findings suggest that TPM2 may be a new therapeutic target for AS and provide relevant evidence for further research on the role of TPM2 in AS.
ACTC1 is mainly involved in the formation of actin fibrous tissue, regulation of myosin binding, positive regulation of gene expression, and negative regulation of apoptosis. Abnormal expression of ACTC1 can be involved in the occurrence and development of many diseases. Ohtaki found that ACTC1 is abnormally expressed in glioma tissues. At the same time, through bioinformatic analysis, Ohtaki et al. (2017) found that ACTC1 is related to the survival of glioma patients, suggesting that ACTC1 may be used as a biomarker for judging the prognosis of glioma. Wanibuchi et al. (2018) found that knocking out the ACTC1 gene can inhibit migration of glioblastoma cells, suggesting that ACTC1 may be a target for glioma treatment. Jiang et al. (2010) found that the decreased expression of ACTC1 may be involved in the occurrence and development of congenital heart disease by inducing the apoptosis of cardiomyocytes. Greenway et al. (2014) also found that ACTC1 is abnormally expressed in patients with congenital heart disease through sequencing, suggesting that it may be a potential therapeutic target. Through bioinformatic analysis, we found that ACTC1 expression is low in patients with AS. We speculate that ACTC1 may be involved in the occurrence and development of AS by inducing apoptosis, affecting actin and myosin function. It may be a therapeutic target. The mechanism of ACTC1 affecting AS is worth further exploration.
PLS3 is mainly involved in affecting calcium ion binding, actin binding, actin filament bundle assembly, and actin filament network formation. The abnormal expression of PLS3 can be involved in the occurrence and development of various diseases. Kampe et al. (2017) found that the low expression of PLS3 may affect bone homeostasis and even lead to severe osteoporosis. Shao et al. (2019) found that the abnormal expression of PLS3 is associated with osteoporotic fractures in postmenopausal women, suggesting that it may be a therapeutic target. Through bioinformatic analysis, Cao et al. (2018) found that PLS3 may be involved in the occurrence and development of gastric carcinoma. Kujawski et al. (2015) found that PLS3 is associated with colorectal carcinoma. Further analysis suggested that PLS3 may be a target for the diagnosis and treatment of colorectal carcinoma. Through bioinformatic analysis, we found that PLS3 expression is low in AS patients. We speculate that PLS3 may be involved in the occurrence and development of AS by affecting the function of actin and cell migration, which may serve as a potential diagnostic and therapeutic target.
CCR2 mainly influences protein binding, blood vessel remodeling, regulation of vascular endothelial growth factor production, and negative regulation of angiogenesis. CCR2 is a key regulator of monocyte and macrophage transport, which can be involved in the occurrence and development of various diseases (Huma et al., 2017). Now, there is much evidence that CCR2 is involved in the occurrence and development of AS. Boring et al. (1998) found that inhibiting the expression of CCR2 can delay the occurrence and development of AS. Weisberg et al. (2006) found that CCR2 can affect inflammation of adipose tissue and insulin resistance. Further analysis suggested that CCR2 may be involved in the occurrence and development of AS by regulating the migration of monocytes and macrophages. Ali et al. (2008) found that the anti-inflammatory effect of endothelial cells mediated by CCR2 can inhibit the progression of AS caused by vascular injury. Verweij et al. (2018) found that the expression of CCR2 in monocytes is associated with arterial wall inflammation in patients with high cardiovascular risk, suggesting that CCR2 may be a therapeutic target for atherosclerotic plaque inflammation. Consistent with the previous research results, through bioinformatic analysis, we found that CCR2 is highly expressed in AS patients.
We hypothesized that CCR2 can promote the occurrence and development of AS by regulating the vascular endothelial growth factor, vascular remodeling, and other pathways. The specific mechanism of the involvement of CCR2 in AS deserves further study. CCR2 may be a therapeutic target for AS.
Despite the rigorous bioinformatic analysis carried out in this study, there are still some shortcomings. First, the amount of data in our study is limited. There may be some deviations in the results. Expanding the sample size can improve the accuracy of the analysis results. Second, we only did confirmatory animal experiments. Although it can be explained to a certain extent that the TPM2 molecule plays an important role in the occurrence and development of AS, there are still differences between animals and humans. Further evidence is still needed to verify the correlation between TPM2 and AS.
5. Conclusion
In conclusion, the present research aimed to identify DEGs that might be involved in the occurrence or development of AS. Finally, 211 DEGs and 18 hub genes were confirmed between atherosclerotic tissues and nonatherosclerotic tissues, which could be used as diagnostic and therapeutic biomarkers for AS. However, the biological functions of all the hub genes in AS require further research.
Footnotes
Acknowledgment
The authors are thankful to Qing-Qing Wang for his statistical assistance and suggestions during the submitting process.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.