
Abstract
The DNA sequencing process has evolved rapidly due to the development of new technologies and equipment capable of producing large amounts of sequencing data. Among these methods, PacBio stands out. The PacBio method uses single molecule real-time, generating sequence files composed by long reads. Storage and analysis of the data generated became a challenge ushering in the development of bioinformatic tools. One of these challenges is the alignment of these sequences. This article describes techniques and processes developed for long DNA sequence alignment using manycore architecture.
1. Introduction
The discovery of the structure of the DNA by Watson and Crick in 1953, based on the crystallography results obtained by Rosalind Franklin and Maurice Wilkins (Watson and Crick, 1953; Zallen, 2003), made possible the conceptual knowledge for DNA replication and proteins encoding, providing the development of methods to sequence the DNA molecule.
The main breakthrough on sequencing technologies occurred in 1977 through Sanger's method, also known as chain-termination or dideoxy technique (Sanger et al., 1977). Improvements on this method, such as detection based on fluorescence through capillary-based electrophoresis, allowed the development of the automated DNA sequencing (Ansorge et al., 1987; Kambara et al., 1988; Swerdlow and Gesteland, 1990), composing the first-generation DNA sequencing platforms.
These first-generation techniques are able to produce reads of about one kilobase (kb) in length. Thus, to increase the length of the fragments to be sequenced and analyzed, the shotgun sequencing was introduced. In this technique, clones of overlapped DNA sequenced fragments are assembled, in silico, into longer contiguous sequences named as contigs.
The possibility to obtain genomic sequences required the use of computational resources in biology, which began in the 1970s. When DNA sequencing processing started, the amount of data obtained grew slowly as a result of the technologies used for sequencing. With the discovery and refinement of new sequencing techniques, the amount of data increased rapidly, requiring high storage and processing capacity. In the past decades the amount of biological data has grown exponentially and researches leading to the development of sophisticated bioinformatics resources made possible the rapid and cost-effective data mining and analyses, approaching the generation of genomics data and its analysis by conventional biological methods (Chu et al., 2017; Costa, 2017).
DNA sequencing is currently in its third generation, for instance the single molecule real-time sequencing platform, developed by Pacific BioSciences, which uses a DNA polymerase anchored in a well small enough to act as a zero-mode waveguide. The polymerase acts on a single integrate DNA molecule incorporating fluorophores labeled nucleotides, which are excited by a laser. The resulting optical signal is recorded by a high-speed camera in real time (Chu et al., 2017).
The more efficient use of the resources, due to the advances in the architecture, made possible the improvement of the computational processing power, memory, and data storage, required by algorithms and alignment programs. There are some alignment algorithms, such as BLASR, DALIGNER, MHAP, Graph-Map, and Minimap, developed for the alignment of long sequences. These programs have different ways of approaching their algorithms in relation to the use of computational resources (Chu et al., 2017).
In this scenario, this study discusses how the techniques for long DNA sequences alignment have been used in a manycore architecture as Intel and NVIDIA.
2. Alignment
In bioinformatics, alignment of sequences is a process to organize and compare DNA sequences to a reference genome/sequence. Alignments can be made between two sequences, known as simple alignment, or among three or more sequences, thus named multiple alignment.
In contrast, alignments can be performed in two different approaches: global alignment or local alignment as seen in Figure 1.
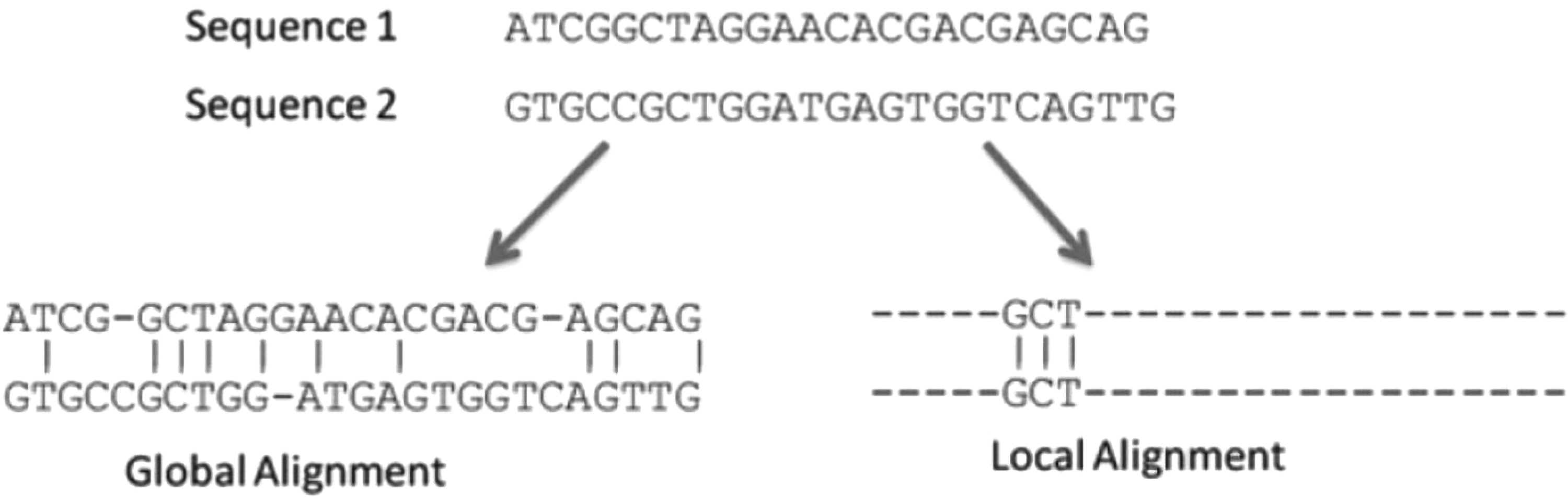
Global and local alignment processes. Note that in global approach both sequences entirely considered, whereas in local alignment only part of the sequences are taken into account for alignment (Prakhar et al., 2018).
2.1. Global alignment
Global alignment is a process in which target DNA sequences are aligned to the reference sequence by each residue. In other words, it takes the entire sequences into consideration, performing an end-to-end alignment. The higher dissimilarity between query and reference, the greater number of gaps will be observed. The main algorithm for this approach is the Needleman–Wunsch, which is based on dynamic programming, developed by Saul Needleman and Christian Wunsch in 1970 (Needleman and Wunsch, 1970).
2.2. Local alignment
Unlike global alignment, local alignments consider subsections of the sequences to match to the reference. This is useful to find regions and motifs with similarity between target and reference sequences. The Smith–Waterman algorithm is a general method for local alignment, also based on dynamic programming, but with different choices to start and end at any place (Smith and Waterman, 1981; Polyanovsky et al., 2011).
3. Manycore Architecture
Parallel systems with high computational power combined to algorithms that perform alignments through parallelized techniques have been widely used to process large amounts of data produced by DNA sequencing platforms. These systems include architectures developed, for example, by Intel and NVIDIA. They are increasingly being used to process this biological information (Cantacessi et al., 2012).
3.1. Intel Xeon Phi architecture
Intel Xeon Phi is a very cost-effective option to be used as a parallel system to perform DNA sequence analyses that has been made available in the recent years. This processor delivers massive parallelism and vectorization focused on high-performance computing, which uses parallel processing for large data demands in different areas, such as computational physics, chemistry, biology, finance, and bioinformatics (Rahman, 2013).
The Knights Landing memory architecture has two types of memory: multichannel DRAM (MCDRAM), a high-bandwidth memory integrated on package, capacity of which is up to 16 GB and peak bandwidth >450 GB/s; and external double-data-rate (DDR), capacity of which is up to 384 GB (64 GB per channel) and peak bandwidth around 90 GB/s.
The Intel Xeon Phi processor supports different types of memory, organizing it into three types of memory mode: (1) flat mode, in which modifications are required to use both the DDR and the MCDRAM in the same application; (2) cache mode, which does not require any software modification and works well in different applications; (3) hybrid mode, which uses part of the MCDRAM as a cache (Codreanu Rodrguez, 2017).
Other important configuration in Knights Landing is interconnecting mesh. There are three clustering modes operating, as follows:
All-to-all: Memory addresses are uniformly distributed across all tag directories in the chip. This is the most general mode with the easiest programming model. Quadrant: In the quadrant clustering mode, which divides the cores into two (hemispheres) or four parts called quadrants and attempts to decrease intraprocess communication time by keeping all threads of a single process close together. Sub-NUMA (sub-nonuniform memory access): Attempts to increase memory performance by keeping shared memory accesses to MCDRAM closer to the quadrant where the request originated. This mode provides the lowest latency, provided that applications are NUMA-aware.
As an example of its application, Intel Xeon Phi was carried out in a multicore architecture for DALIGNR software, developed to align long sequences (Myers, 2014). DALIGNER had a better performance being executed on Xeon Phi when compared with a conventional multicore processor. Tuning the Intel Xeon Phi architecture parameters was observed an improvement of 14% over the results compared with a conventional multicore processor (Costa et al., 2019).
In addition, in 2014 the development of the SWAPHI and SWAPHI-LS programs, making use of Intel Xeon Phi coprocessors, made possible the acceleration of the Smith–Waterman algorithm for protein database searching and for long DNA sequences alignment, respectively. The Smith–Waterman algorithm is a well-known algorithm for performing local sequence alignment; that is, for determining similar regions between two sequences (Yongchao and Bertil, 2014; Yongchao et al., 2015).
3.2. GPU architecture
Developed by NVIDIA in the 1990s, the graphical processing units (GPU) use many cores for parallel mass processing, focused on energy efficiency and throughput improvement. Initially developed to meet the needs of the gaming area, GPU quickly proved to be very efficient also in other areas, such as engineering, medicine, economics, and finance (Zanotto et al., 2019).
Currently, the computational power of a GPU is higher than the computational power of the central process unit (CPU), due to the fact that CPUs are composed by a few cores that can handle a few software threads at a time, whereas GPUs are composed by hundreds of cores able to handle thousands of threads simultaneously.
Aiming to explore the capabilities of the GPU in an easy and straightforward way, in 2006 NVIDIA developed a parallel computing platform and programming model called compute unified device architecture (CUDA), able to support various languages and programming environments (Zanotto et al., 2019).
In face of CUDA's processing power, in 2010 the program CUDAlign was developed aiming to align long DNA sequences making use of the Smith–Waterman algorithm in a GPU architecture. CUDAlign algorithm is able to compare megabase biological sequences with an exact Smith–Waterman affine gap variant. It was also implemented in CUDA, testing in two GPU boards separately. Later in 2014 a new version of CUDAlign (v.3.0) improved its efficiency executing the Smith–Waterman algorithm in a parallel strategy, on multi-GPU platforms, to the alignment of long DNA sequences (Sandes et al., 2014)
Taking advantage of the CUDA's efficiency, in 2009 CUDASW was developed optimizing Smith–Waterman protein database searches. The CUDASW is a fast Smith–Waterman protein database search algorithm, which couples CPU and GPU SIMD (single instruction, multiple data) instructions and carries out concurrent CPU and GPU computations (Liu et al., 2013).
Multiple sequence alignments (MSA) were also improved by this interesting computer strategy through the development of the MSACUDA program, which provides high performance for MSA alignments. MSA-CUDA parallelizes all three stages of the ClustalW processing pipeline using CUDA and achieves significant speedups compared with the sequential ClustalW program for a variety of large protein sequence data sets (Liu et al., 2009).
4. Conclusion
We presented in this study the progress of bioinformatic tools to provide the necessary resources for DNA sequences alignment, whether local or global approach. Currently, as long reads sequencing has become widely required, the application of manycore architectures, such as Intel Xeon Phi and NVIDIA GPU, and the development of more efficient algorithms have considerably improved the DNA sequences alignment process. This is a dynamic area and NVIDIA GPU architecture stands out for the rapid development of new devices with a high processing power.
Footnotes
Acknowledgments
The authors thank the department of computer science, University of the Federal of Rio de Janeiro, for providing the computational resources used to carry out the experiments presented in this study.
Author Disclosure Statement
The authors declare they have no competing financial interests.
Funding Information
The authors received no funding for this article.