
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal malignancies worldwide due to its ineffective diagnosis and poor prognosis. It is essential to identify differentially expressed genes (DEGs) in PDAC to gain new insights into its underlying molecular mechanisms, as well as identify potential diagnostic and therapeutic targets. We screened 135 DEGs from the GSE15417, GSE16515, and GSE28735 PDAC and normal pancreatic tissue microarray data sets, and identified 16 DEGs that were correlated with PDAC prognosis through the Kaplan–Meier survival analysis and log-rank tests. The Cancer Genome Atlas and Oncomine databases validated the expression levels of 16 candidate genes (SLC6A14, GPRC5A, IFI27, ERP27, SDR16C5, SIDT2, TCN1, COL12A1, MMP1, CEACAM6, DKK1, ITGA2, KRT19, PLAU, ANO1, and GABRP). Weighted gene coexpression network analysis (WGCNA) and protein and protein interaction (PPI) analysis identified three hub genes—ERP27, ITGA2, and MMP1—that are likely important in PDAC prognosis. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis demonstrated that they were enriched in functions of extracellular matrix organization, extracellular structure organization, and positive regulation of cell migration. Taken together, we identified three pivotal genes for PDAC, which can improve our understanding of its pathogenesis, progression, and prognosis.
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal malignancies on account of its ineffective diagnosis and therapy (Oncology, 2014). The median survival duration of PDAC is less than 6 months, and the 5-year overall survival (OS) rate is a dismal 7.7% (Pierce, 2012; Oncology, 2014; andrew_aguirre@dfci.harvard.edu, 2017). Although early detection and surgical resection have increased the 5-year survival rate of stage I PDAC patients to 50%, most patients are already at the advanced stage when diagnosed (Hernandez and Lucas, 2016; Li et al., 2016; Taucher et al., 2016). Therefore, it is crucial to identify diagnostic or prognostic biomarkers and therapeutic targets to improve clinical outcomes.
Several mutations drive the development and progression of PDAC (Dickson, 2017), and understanding these genetic alterations in the context of biological pathways can help identify specific novel biomarkers. Zhou et al. (2015) demonstrated that p21-activated kinase 1 (PAK1) regulated the downstream signaling pathways of multiple growth factors, such as hepatocyte growth factor and MET receptor tyrosine kinase. Recent studies indicate that KRAS, SMAD4, CDK6, SOX9, GATA6, PIK3CA, and PIK3R3 are frequently mutated in PDAC (Fakhri and Lim, 2017). The mutagenic landscape of PDAC also includes genes related to histone modification, DNA damage repair, and mitotic cytokinesis, as well as drug targets such as ERBB2, MET, and FGFR1 (Dunne and Hezel, 2015; Long et al., 2016; Zhou et al., 2018).
High-throughput gene profiling and sequencing technologies have helped identify cancer-associated genes and biological pathways on the basis of histological tumor classification and clinical data. The Cancer Genome Atlas (TCGA) database includes the gene expression data of almost 30 human cancers (Jia et al., 2018), and the Gene Expression Omnibus (GEO) is a public database of microarrays or gene profiles of a wide range of diseases, including cancer (Edgar and Lash, 2002). The weighted gene coexpression network analysis (WGCNA) helps identify the correlation patterns among genes across microarray samples, and gene set enrichment analysis (GSEA) is a computational method that identifies the significance of a gene set vis-a-vis different physiological states (Holden et al., 2008). These bioinformatic resources have unearthed several novel biomarkers for the diagnosis, treatment, and prognosis of different cancer types, essential for the development of individualized treatment strategies.
The aim of this study was to systematically review all available gene expression data for PDAC, and perform a pooled analysis using the above bioinformatic tools to identify biomarkers and molecular targets. We identified ERP27, ITGA2, and MMP1 as novel potential diagnostic and prognostic biomarkers for PDAC, which need to be validated further for clinical application.
2. Materials and Methods
2.1. Patient samples
The study included two PDAC patients recruited from Shenzhen People's Hospital. Written informed consent was obtained from all patients before surgery, and the study was approved by our Institutional Ethics Committee.
2.2. Microarray data and identification of differentially expressed genes
The GSE15471, GSE16515, and GSE28735 microarray data sets—consisting of gene expression data of 117 PDAC and 97 adjacent nontumor tissues—were obtained from the GEO. The raw data of each microarray were normalized and analyzed by R package, limma, and RobustRankAggreg package. Differentially expressed genes (DEGs) were identified by classical t test with false discovery rate <0.05 and | logFC | >1 as the cutoff criterion.
2.3. Gene Ontology, Kyoto Encyclopedia of Genes and Genomes pathway enrichment, and GSEA
To indentify the potential functions of the identified DEGs, Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and GSEA were performed by R cluster Profiler package as previously described (Yu et al., 2012).
2.4. WGCNA and protein and protein interaction network analysis
The DEGs were qualified and then used to construct a WGCNA network by R WGCNA package. The primary protein and protein interaction (PPI) network was constructed by module node >0.6, and uploaded onto the Cytoscape software (www.cytoscape.org/), which was used to construct PPI subnetworks and obtain candidate genes by calculating node degree.
2.5. OS analysis
To analyze the prognostic value of the candidate genes, the PDAC patients were stratified into the high-expression and low-expression groups for each DEG, using the median expression level from TCGA (https://cancergenome.nih.gov/) as the cutoff. The two patient cohorts for each DEG were compared by Kaplan–Meier analysis and the log-rank test using the R survival package.
2.6. Analysis of candidate gene expression levels
The expression of candidate genes was analyzed by FirebrowseR package for PDAC from TCGA, which offers customizable functions such as tumor/normal tissue differential expression analysis, profiling according to cancer types or pathological stages, patient survival analysis, similar gene detection, correlation analysis, and dimensionality reduction analysis. The expression data were further validated using Oncomine (https://www.oncomine.org), a web application for translational bioinformatics that can compute gene expression signatures, clusters, and gene set modules, and automatically extracts biological insights from the raw data.
2.7. Identification of the candidate gene protein expression
The expression of the proteins encoded by the candidate genes was identified from the Human Protein Atlas (HPA; www.proteinatlas.org/), which includes quantitative transcriptomic data (RNA-Seq) and spatial proteomic data from immunohistochemistry (IHC) on tissue microarrays.
2.8. Immunohistochemistry
The expression of relative proteins can be analyzed by IHC. First, tissue slices were incubated overnight at 4°C with primary antibodies to ERP27, ITGA2 and MMP1. Then they were incubated with secondary antibodies at room temperature for 1 hour. Finally, slides were stained with DAB, counterstained with haematoxylin and photographed.
2.9. Statistical analysis
The data are presented as mean ± standard deviation. Dependent t test was used to compare the mean between two groups. Figure 5 shows the log-rank tests used.
3. Results
3.1. Identification of DEGs
Three microarray data sets of pancreatic cancer and adjacent nontumor tissues—GSE15471, GSE16515, and GSE28735—were downloaded from the GEO database and analyzed. GSE15471 and GSE28735 contained data from 36 PDAC and 45 paired adjacent nontumor tissues, and GSE1615 from 36 PDAC and 16 adjacent nontumor tissues, altogether comprising 117 PDAC and 97 nontumor tissues. A total of 135 DEGs were identified, of which 86 were upregulated and 49 downregulated in the PDAC compared with the normal tissues (Table 1). The top 20 DEGs are shown in Figure 1.
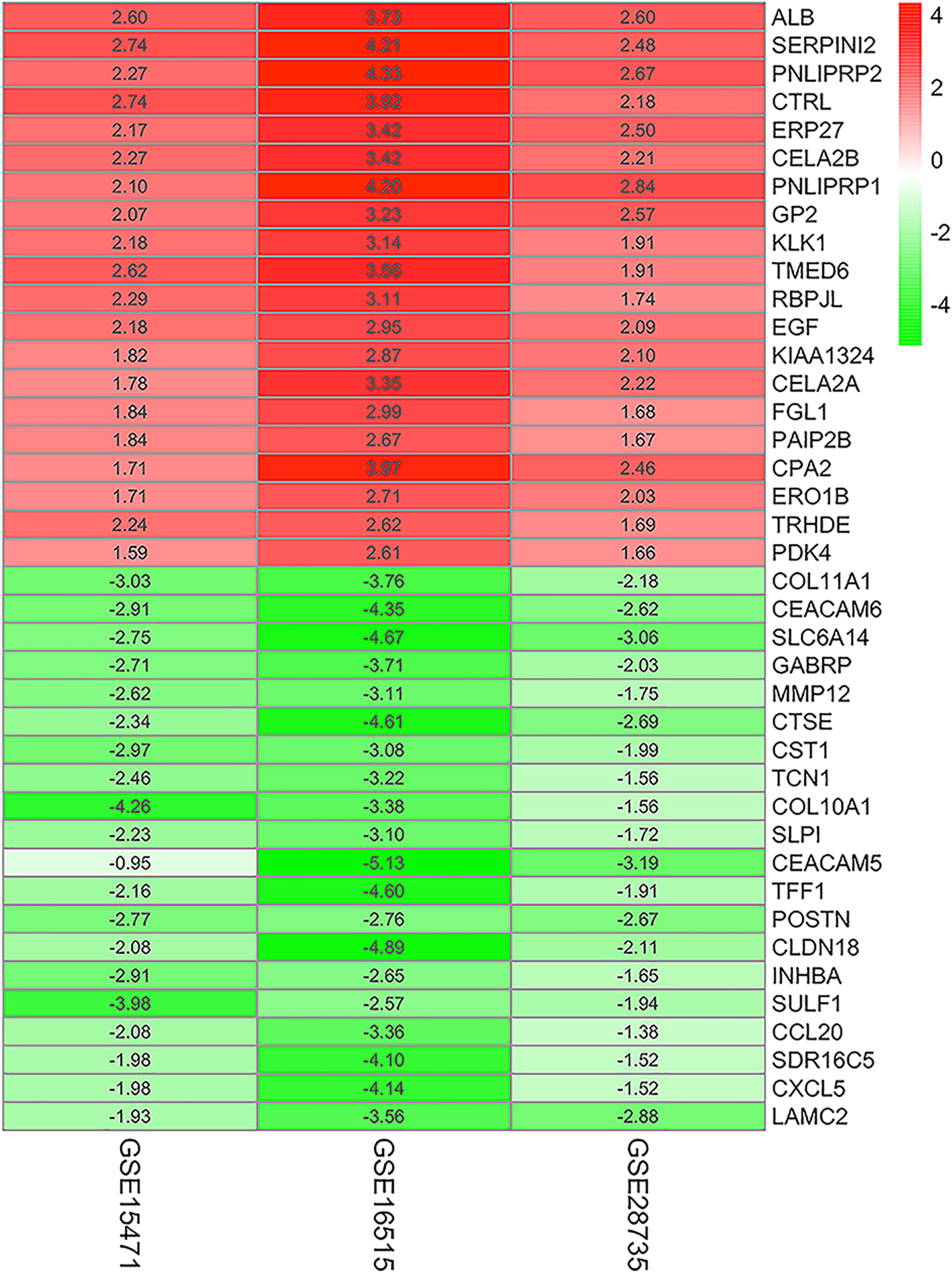
The DEGs in PDAC GSE15417, GSE16515, and GSE28735 were obtained and calculated by R package, and then indentified DEGs as FDR <0.05 and |FC| >1 as cutoff. The top 20 DEGs are shown in the figure, and red/blue were relative upregulation/downregulation comparing adjacent nontumor tissues with PDAC tissues. DEGs, differentially expressed genes; FDR, false discovery rate; PDAC, pancreatic ductal adenocarcinoma.
The Differentially Expressed Genes in Pancreatic Ductal Adenocarcinoma
DEGs, differentially expressed genes.
3.2. Functional enrichment analysis
The DEGs showed significant enrichment in the biological process (BP) terms of leukocyte migration, digestion, and extracellular matrix (ECM) organization, the cellular component (CC) terms proteinaceous ECM, endoplasmic reticulum lumen, and secretory granule lumen, and the molecular function (MF) terms endopeptidase activity, serine-type endopeptidase activity, serine-type peptidase activity, and serine hydrolase activity (Fig. 2A). It was displayed that most significant genes were involved in significant biological function, for instance, MMP1 may regulate digestion, multicellular organismal catabolic process, ECM organization, extracellular structure organization, and leukocyte (Fig. 2B). It was displayed that the gene network may regulate the biological function, such as FN1, ITGA2, LAMC2, CEACAM5, CXCL5, OLR1, and DKK1 regulating positive regulation of cell migration (Fig. 2C). Core enriched genes in metabolic pathways for KEGG pathway were discovered (Fig. 2D). Running enrichment score was the traditional method for visualizing GSEA. The results showed significant gene enrichment in pancreatic secretion (Fig. 2E).
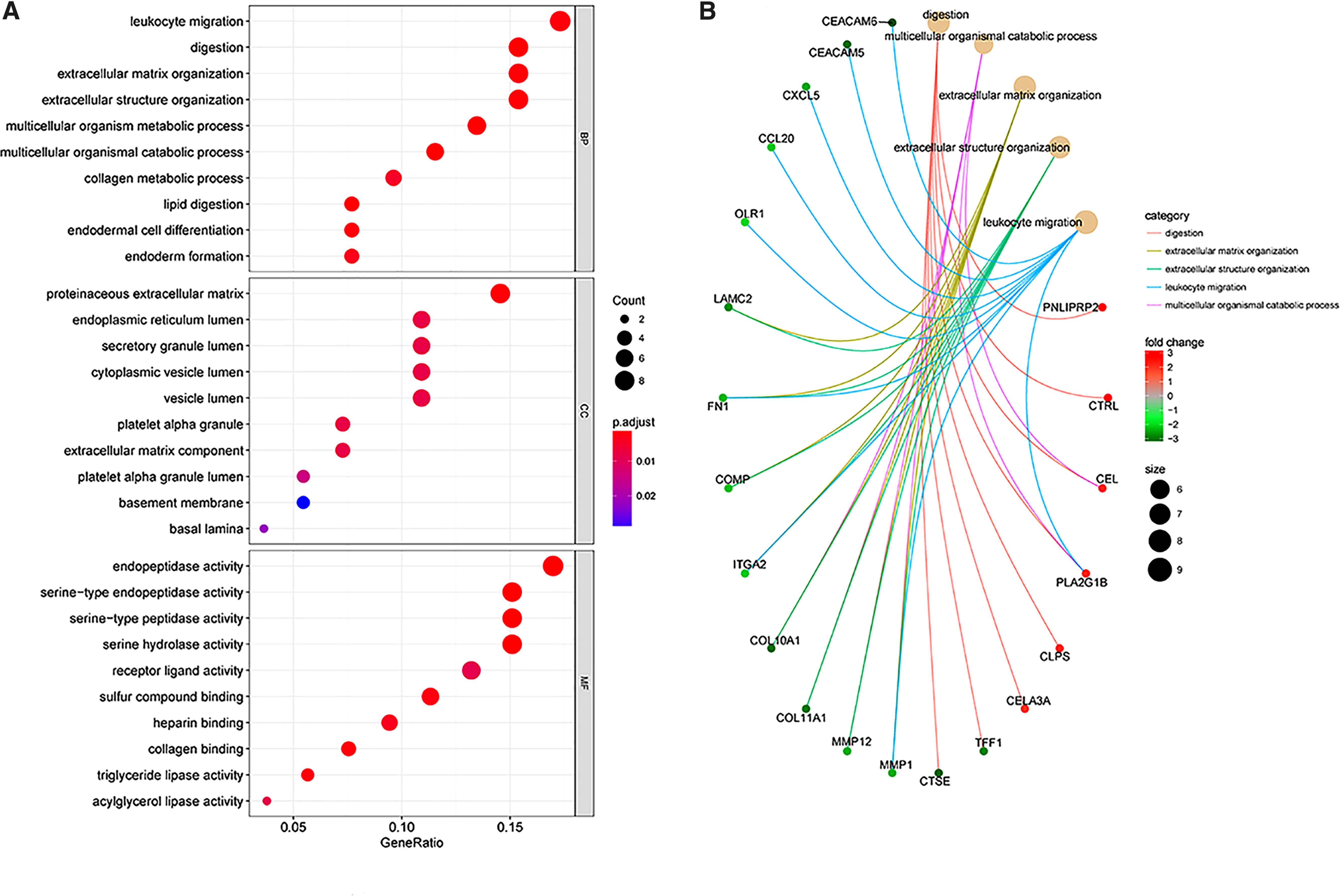
GO and Pathway analysis of the DEGs.
3.3. Survival analysis for candidate genes
The PDAC patients were stratified into the high-expressing (n = 86) and the low-expressing (n = 49) groups for the 135 DEGs, using the median expression level as the cutoff, and the survival was analyzed by R package (data not shown). Sixteen DEGs were correlated with PDAC prognosis, with high expression of 16 of them—ITGA2, MMP1, ERP27, ANO1, COL12A1, DKK1, EPYC, GABRP, KRT19, PLAU, CEACAM6, GPRC5A, IFI27, SDR16C5, SLC6A14, and TCN1—significantly associated with shorter survival (p = 0.001, p = 0.027, p = 0.036, p = 0.002, p = 0.004, p = 0.014, p = 0.005, p = 0.027, p = 0.004, p = 0.002, p = 0.018, p < 0.001, p = 0.003, p = 0.0005, p = 0.005, and p = 0.021, respectively); in contrast, higher SIDT2 expression was associated with longer survival (p = 0.04) (Fig. 3). Therefore, these 17 DEGs were further analyzed.

Survival analysis for DEGs. The DEGs subjected to survival analysis by R package. Kaplan–Meier survival analysis and log-rank tests showed that higher ITGA2, MMP1, ERP27, ANO1, COL12A1, DKK1, EPYC, GABRP, KRT19, PLAU, CEACAM6, GPRC5A, IFI27, SDR16C5, SLC6A14, and TCN1 expression was associated with shorter survival time (p = 0.001, p = 0.027, p = 0.036, p = 0.002, p = 0.004, p = 0.014, p = 0.005, p = 0.027, p = 0.004, p = 0.002, p = 0.018, p < 0.001, p = 0.003, p = 0.0005, p = 0.005, and p = 0.021, respectively), however, the higher SIDT2 expression had longer survival time (p = 0.04).
3.4. DEG expression in PDAC
The mRNA expression data from 178 PDAC and 4 normal tissues were downloaded from TGCA, and the expression levels of the above candidate genes were compared using the R package. ERP27 and SID2 were downregulated (p = 0.03 and p < 0.01, respectively), and CEACAM6, GPRC5, IFI27, SDR16C5, SLC6A14, and TCN1 were upregulated in PDAC (p < 0.01) (Fig. 4A). ANO1, COL12A1, DKK1, GABRP, ITGA2, KRT19, MMP1, and PLAU were overexpressed in normal tissues (p < 0.01), while EPYC levels were similar in both PDAC and normal tissues (p > 0.05) (Fig. 4B). Oncomine database validated the findings of TGCA, and the results demonstrated that ERP27 and SID2 were underexpressed in PDAC (p < 0.01), but ITGA2, MMP1, CEACAM6, GPRC5, IFI27, SDR16C5, SLC6A14, TCN1, ANO1, COL12A1, DKK1, GABRP, KRT19, EPYC, and PLAU were overexpressed in PDAC (p < 0.01) (Fig. 5). Taken together, the expression of all candidate genes except egg yolk phosphatidylcholine (EYPC) in TCGA and Oncomine was consistent with the gene expression profiles from GEO.
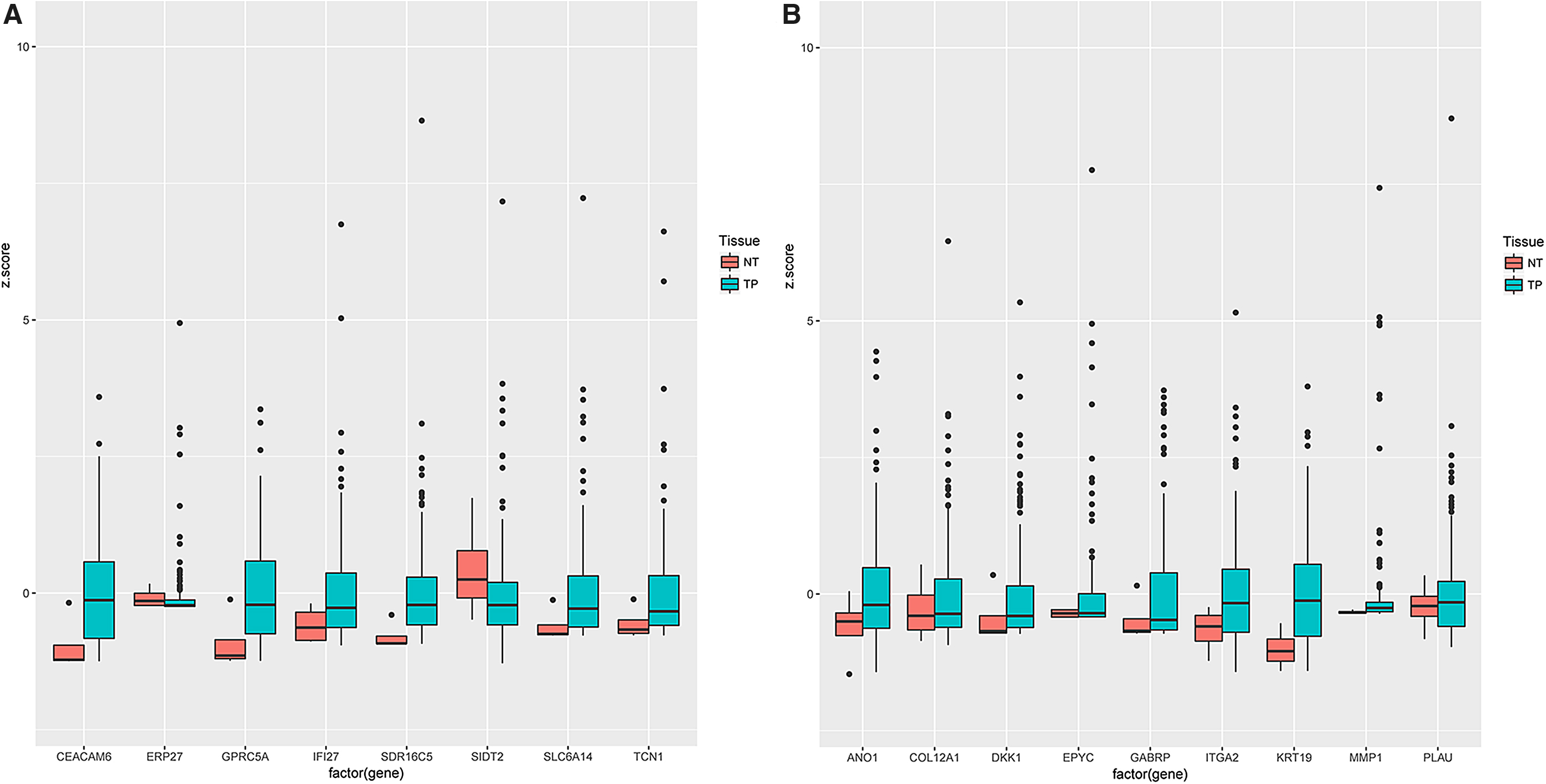
The candidate gene expression in TCGA data. The candidate genes expression was performed by R package from TCGA data.
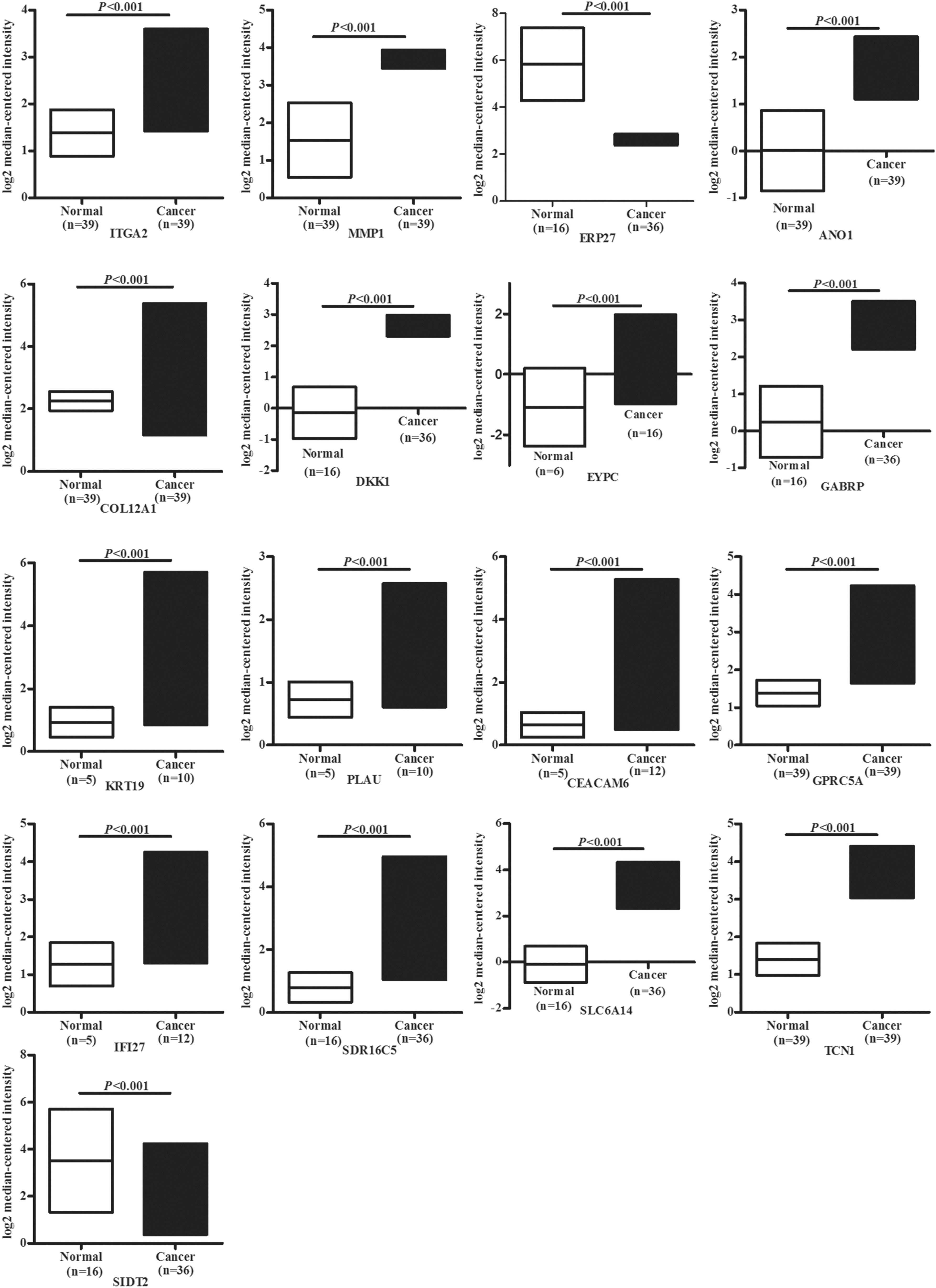
The candidate gene expression in Oncomine data set. The results showed that ERP27 and SID2 were underexpressed in PDAC (p < 0.01); however, ITGA2, MMP1, CEACAM6, GPRC5, IFI27, SDR16C5, SLC6A14, TCN1, ANO1, COL12A1, DKK1, GABRP, KRT19, EPYC, and PLAU were overexpressed in PDAC (p < 0.01).
3.5. WGCNA and PPI network analysis and candidate gene identification
DEGs with similar expression patterns were clustered by WGCNA, which divided them into three modules: gray, turquoise, and blue (Fig. 6A) (Supplementary Table S1), and the PPI network was then constructed with 0.6 as the threshold. Further PPI subnetworks were constructed using Cytoscape, resulting in 39 nodes and 237 edges. Combined with the survival analysis, we obtained three core candidate genes—ITGA2, MMP1, and ERP27—on the basis of node degrees. The red and blue represent the up- and downregulation, respectively, and the degree is displayed with the size (Fig. 6B).
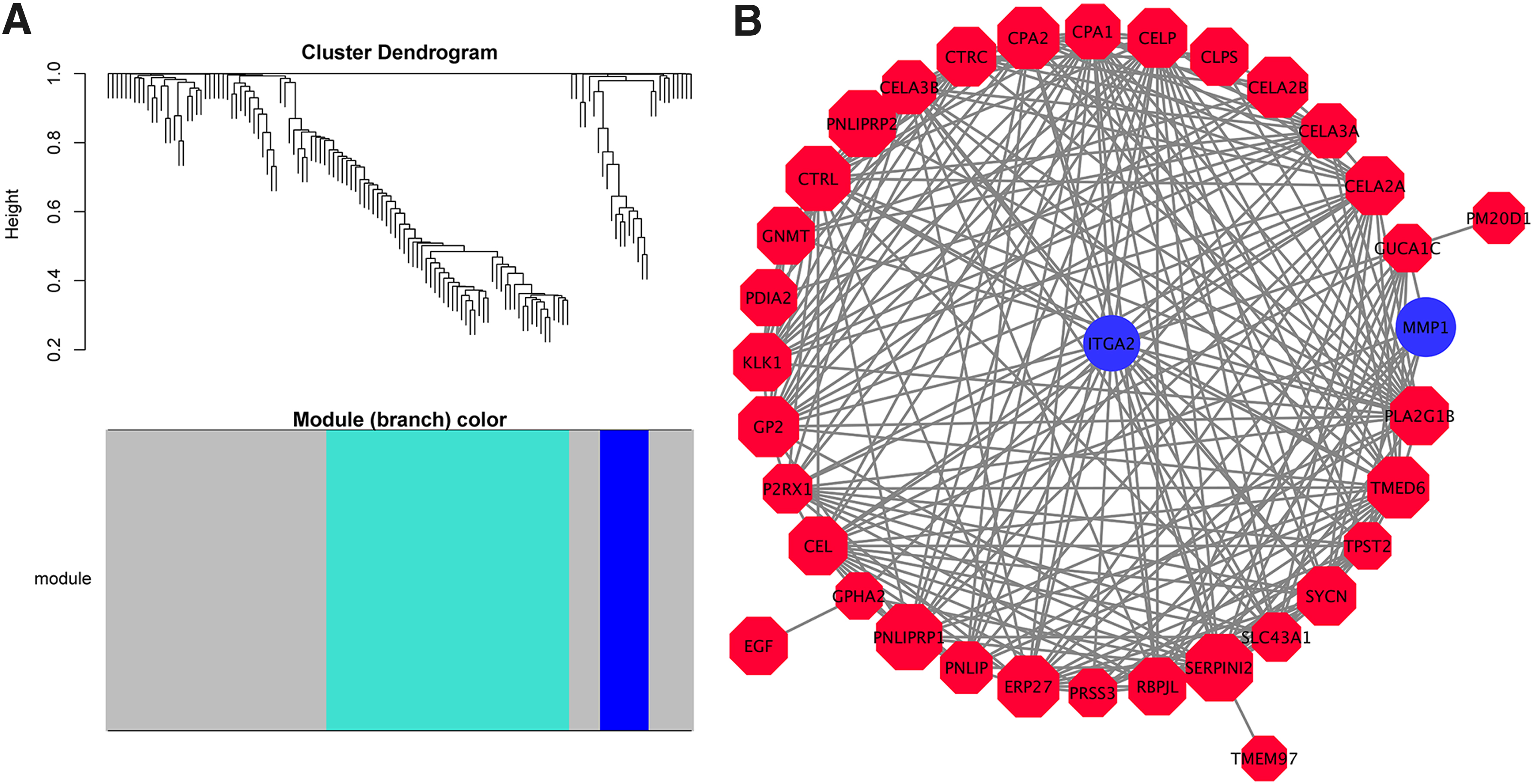
WGCNA and PPI analysis and candidate gene identification.
3.6. Candidate gene protein expression validation
The IHC and patient clinical data were extracted from HPA, which showed higher in situ expression of ITGA2 and MMP1 in PDAC tissues compared with normal pancreatic tissues, while ERP27 expression was lower in the PDAC tissues (Fig. 7). We have tested ITGA2, MMP1, and ERP27 protein expression in two paired PDAC tissues by IHC, and the results showed a higher ITGA2 and MMP1 expression in PDAC tissues compared with normal pancreatic tissues; yet, ERP27 expression was lower in the PDAC tissues (Supplementary Fig. S1).

Candidate gene protein expression validation. The IHC data were obtained from HPA. Staining yielded ITGA2, and MMP1 protein expression was higher in PDAC tissues than in normal pancreatic tissues, but ERP27 protein expression was lower in PDAC tissues than in normal pancreatic tissues. HPA, Human Protein Atlas; IHC, immunohistochemistry.
4. Discussion
PDAC has one of the worst survival rates among all cancers. Over the last decade, comparative genome-wide analyses has identified several loci associated with a high susceptibility to PDAC (Bhasin et al., 2015; Kong et al., 2016; Makohon-Moore et al., 2017; Ghosh et al., 2018). In the present study, we identified 135 DEGs from the GSE15471, GSE16515, and GSE28735 data sets of PDAC and normal pancreatic tissue microarrays, which minimized the statistical deviation (Zhou et al., 2018). To further extract the tumor-related genes from the DEGs with greater accuracy, we analyzed the survival trends of the PDAC patients relative to the DEG expression levels, and found that 16 genes were negatively correlated and only 1 gene was positively correlated with survival. Of these 17 genes (SLC6A14, GPRC5A, IFI27, ERP27, SDR16C5, SIDT2, TCN1, COL12A1, EPYC, MMP1, CEACAM6, DKK1, ITGA2, KRT19, PLAU, ANO1, and GABRP), all except EPYC were validated by TGCA as candidate genes likely playing a pivotal role in PDAC development. Seven of these candidate genes (SLC6A14, GPRC5A, ANO1, GABRP, ITGA2, KRT19, and PLAU) have been previously associated with PDAC.
In addition, ANO1, ITGA2, and KRT19 are well-established oncogenes (Ischenko et al., 2013; Liu et al., 2015; Nones et al., 2014; Sauter et al., 2015; Sauter et al., 2015a, 2015b; Yao et al., 2016), and genetic analysis has further reinforced the association between PLAU and PDAC (Bournet et al., 2012; Krug et al., 2014; Gutiérrez et al., 2015). A previous study showed that SLC6A14, GPRC5A, and GABRP were suitable therapeutic biomarkers for PDAC (Karunakaran et al., 2010; Mccann, 2015; Nakamura, 2008; Penheiter et al., 2015; Takehara et al. 2007). In this study, we identified some additional oncogenes—namely IFI27, ERP27, SDR16C5, SIDT2, TCN1, and COL12A1—that have not been previously linked to PDAC.
According to GO and KEGG pathway analyses, the most significantly enriched CC, MF, and BP terms among the oncogenes are proteinaceous ECM, endopeptidase, and serine-type endopeptidase activity, and positive regulation of leukocyte migration and ECM organization, respectively. This is highly significant since the ECM is a major factor influencing cancer metastasis, and PDAC in particular is highly invasive and metastatic (He et al., 2013). Furthermore, pancreatic secretion and digestion are now considered an important oncogenic function in the gastrointestinal cancers, and aberrant expression of genes that regulate ECM organization and pancreatic secretion has been associated with aggressive pancreatic tumors (Laklai et al., 2012; He et al., 2013; Biondani et al., 2018).
The major hub genes involved in PDAC were further screened by combining GSEA, WGCNA, PPI networks and survival analysis, which identified MMP1, ITGA2, and ERP27. All three genes were enriched in ECM organization and extracellular structure functions. MMP1 and ITGA2 were overexpressed, while ERP27 was inhibited in the PDAC tissues. Matrix metalloproteinase 1 (MMP1) gene is part of a cluster of MMP genes on chromosome 11 that encode enzymes such as collagenase-1, fibroblast collagenase, and interstitial collagenase. MMP1 is the key mediator in the breakdown of ECM, with major roles in embryogenesis, propagation, and tissue remodeling in various physiological processes. MMP1 also has a significant impact on cancer metastasis, since the degradation of basal membranes and ECM enables cancer cell migration and invasion. MMP1 overexpression is associated with the metastasis of various cancers, such as breast cancer, lung carcinoma, and colorectal cancer (Sunami et al., 2000; Hinoda et al., 2002; Zinzindohoué et al., 2005; Decock et al., 2008; Sauter et al., 2008).
Integrin α2 (ITGA2) gene is located on chromosome 5 and encodes the transmembrane receptor for collagens and related proteins. It is responsible for the adhesion of platelets and other cell types to the ECM (Ji et al., 2011; Heino, 2012). Nones et al. (2014) demonstrated upregulation of ITGA2 in 167 untreated resected PDAC compared with 29 adjacent nontransformed pancreatic tissues. In addition, ITGA2 mRNA levels are significantly high in gastric cancer tissues, and blocking ITGA2 not only inhibited cell migration but also induced apoptosis in gastric cancer cells (Chen et al., 2011). Xu et al. (2017) reported that ITGA2 stimulated yes-associated protein (YAP) activity and was associated with poor survival of PDAC patients. Furthermore, Zhao et al. (2015) found that ITGA2 was also a key gene involved in hepatocellular carcinoma (HCC) development.
ERP27 encodes a redox-inactive member of the protein-disulfide isomerase family, which assists almost one-third of all cellular proteins that pass and undergo oxidative folding in the endoplasmic reticulum, indicating its function in regulating pancreatic secretion (Alanen et al., 2006). Transcriptome analysis showed higher expression of ERP27 in the exocrine glandular cells compared with other tissues, which decreased significantly in an acute pancreatic model (Chen et al., 2010; Kober et al., 2013).
Taken together, we identified three pivotal genes that are potentially involved in PDAC prognosis through bioinformatic analysis, greatly contributing to our understanding of the pathogenesis, progression, and prognosis of PDAC. Further studies are needed to validate these genes functionally and elucidate their underlying molecular mechanisms.
Footnotes
Acknowledgment
The authors thank Prof. Guangchuang Yu for the use of R package.
Availability of Data and Materials
The data sets used during the present study are available from the corresponding authors on reasonable request. Study concept and design: L.-s.W., J.Y., and D.-f.L. Acquisition of data: X.C. and M.-f.Y. Analysis of data: D.-f.L. and M.-f.Y. Drafting of the article: D.-f.L. Statistical analysis: X.C. Obtained funding: J.Y. and D.-f.L. Revision of the article: W.F.
Ethics Approval and Consent to Participate
Shenzhen People's Hospital Institutional Ethics Committee.
Consent for Publication
The patients provided consent for data to be published. All authors gave approval for publication.
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This study was supported by the Natural Science Foundation of Hunan Province (No. 2017JJ3270); the Natural Science Foundation of Guangdong Province (No.2018A0303100024); the Three Engineering Training Funds in Shenzhen (No. SYLY201718 and ![]() ), and the Technical Research and Development Project of Shenzhen (No. JCYJ20150403101028164).
), and the Technical Research and Development Project of Shenzhen (No. JCYJ20150403101028164).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.