
Abstract
Genome-wide gene expression data for cell model of Parkinson's disease (PD) have considerably improved our understanding of the underlying molecular mechanisms involved in cell death during PD neurodegeneration. Apomorphine (APOM), a nonselective dopamine agonist, has been used to treat patients with advanced PD showing no response to levodopa or other dopamine agonists. Although APOM plays a role as a free radical scavenger with neuroprotective effect, it has been reported that long-term use of APOM in PD treatment brings about side effects such as nausea and orthostatic hypotension. For safe use of APOM in PD treatment, it is crucial to understand the underlying molecular mechanisms of APOM in PD. In this study, two groups of microarray data including PD cell model and APOM added PD cell model were used to survey mediation effects of APOM in PD cell model. Mediation analysis between disease genes obtained from PD cell model and drug genes obtained from APOM added PD cell model was carried out with shortest path model on KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways and partial least squares structure equation modeling. Our results suggest that drug genes responding to APOM might contribute to negative regulation of disease genes by direct or indirect ways.
1. Introduction
Parkinson's disease (PD), the second most prevalent neurodegenerative disease worldwide, is characterized by motor symptoms such as tremor at rest, bradykinesia, rigidity, and postural instability (Sui et al., 2019). Previous studies have demonstrated extensive loss of dopaminergic neurons in the substantia nigra when PD is diagnosed clinically after developing motor dysfunction (Tolosa et al., 2009). Therefore, increase of dopamine in the brain has been used as a basic strategy for treatment of initial PD. Supply of dopamine to the brain can be carried out by levodopa (
Despite the beneficial effect of APOM on patients with advanced PD, its molecular mechanism at cellular level remains unclear. In addition, prolonged use of APOM might result in side effects such as orthostatic hypotension, nausea, and fibrotic nodules at the site of APOM injection (Deleu et al., 2004). For safe use of APOM in PD patients, it is necessary to understand the underlying molecular mechanisms of APOM in PD. Genome-wide gene expressions in PD cell model and APOM-treated PD cell model have been investigated with a commercial whole-genome microarray (human HT-12 expression v.4 bead array; Illumina, Inc.) in our previous studies (Kim et al., 2013; Do, 2017). The PD cell model used in those studies was human neuroblastoma SH-SY5Y cells treated with 1-methyl-4-phenyl-pyridium (MPP+) that could mimic many aspects of dopaminergic (DAergic) neuronal death observed in PD (Choi et al., 2014; Do, 2014). The aim of this study was to determine the molecular function of APOM in PD cell model by analyzing interactions of disease genes related to PD and drug (APOM) genes through the shortest path model on KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways and partial least squares structure equation modeling (PLS-SEM) (Pepe and Burzykowski, 2017). For this, two groups of time-series microarray data, that is, MPP+ treatment group and MPP+ + APOM treatment group, were used from our previous studies (Kim et al., 2013; Do, 2017).
As MPP+-treated SH-SY5Y cells have been extensively used as PD cell model, their gene expression profiles can be used for detecting PD-associated genes. In a similar way, gene expression profiles of MPP+-treated SH-SY5Y cells with APOM were able to discover genes associated with function of the drug (APOM). Expression values for these two groups of genes, that is, disease-associated genes (disease genes) and drug-associated genes (drug genes), were utilized to explore the role of APOM in PD cell model. Number of disease genes and drug genes were 808 and 935, respectively. The number of common genes shared by the two groups was 175. Among these 175 common genes, expression profiles of 47 genes were directly affected by APOM. The analysis of shortest paths between disease-specific and drug (APOM)-specific genes resulted in the identification of 948 mediator genes linking them. These mediator genes might be associated with the underlying molecular mechanism or cellular process of the relationship between disease-specific and drug (APOM)-specific genes. Indirect or mediated effects of mediator genes were tested by PLS-SEM known to be suitable for the analysis of mediating variables.
Results of PLS-SEM mediation analysis suggested that if global expression level of drug genes increased, global expression level of disease genes decreased. Direct effect, indirect effect, and total effect of drug genes on disease genes were −0.26, −0.35, and −0.61, respectively. The indirect effect was carried out by mediator genes, many of them were involved in pathways including ras/dopaminergic synapse/oxytocin/insulin/apelin/sphingolipid signaling pathways. This indicates that drug-specific genes might regulate disease-specific genes through various signaling pathways. Our result might contribute to the understanding of molecular mechanisms of APOM in PD patients.
2. Methods
2.1. Identification of disease and drug genes from time course microarray data
Two groups of time-series microarray data (i.e., gene expression data for MPP+-treated SH-SY5Y cells without APOM and with APOM) were used from the study of Kim et al. (2013) and Do (2017), respectively. To find genes showing significant temporal expression changes in the time-series microarray data, Bioconductor package maSigPro (Conesa et al., 2006) was used. This program identifies differentially expressed genes from time-series microarray data through a two-step regression approach including adjustment of a global model and selection of genes showing significantly different profile. Analysis of single- and multi-series time course microarray data is available in maSigPro. The analysis of single-series time course was carried out to find genes with significant temporal expression changes for MPP+-treated SH-SY5Y cells without APOM and with APOM, respectively. Analysis of multi-series time course was also carried out to identify genes with significantly different profiles between MPP+-treated SH-SY5Y cells without APOM and with APOM.
2.2. Shortest path analysis between drug-specific and disease-specific genes
Genes showing significant temporal expression in MPP+-treated SH-SY5Y cells can be considered as disease genes because these cells have been utilized as a cell model for PD (Do, 2016). On the contrary, genes with significant temporal expression in MPP+-treated SH-SY5Y cells with APOM can be regarded as drug genes because APOM has a neuroprotective effect on patients with advanced PD (De Gaspari et al., 2006; Antonini et al., 2011; Carron et al., 2011). Common genes between disease genes and drug genes can be considered as disease genes on which APOM act directly or as disease genes on which the drug does not have any effect. Drug-specific and disease-specific genes represent disease genes and drug genes except for those common genes, respectively. Indirect association between drug-specific and disease-specific genes could be understood by inspecting for connections between these two groups of genes. The assumption behind the search is that drug-specific genes can act on disease genes. The reason is that treatment with a drug, that is, APOM, should have an effect on disease genes directly or indirectly by other genes.
All KEGG pathways were joined in a unique graph using R packages graphite and KEGGgraph. Direct shortest paths on the KEGG graph between each any drug-specific genes and disease-specific genes were discovered by using R package igraph. To evaluate if the found shortest paths could change significantly with time for any gene that receives a connection in the shortest path, two different equations were generated as illustrated hereunder:
where Gi is the ith gene influenced by the nth gene Gn; βin is the path coefficient between Gi and Gn; T is the time and γi the path coefficient between Gi and T; Ei is the term that encloses the unexplained part in the model of Gi. The difference in the representation of the gene Gi between the two equations is in the consideration of time T in one model. Every equation of the shortest path constitutes the shortest path model. All models generated for a specific shortest path are fitted by SEM as in our previous studies (Pepe and Grassi, 2014; Pepe and Do, 2015, 2016).
To evaluate if time had an effect on the shortest path, a hypothesis test was performed to compare model-implied covariance matrices of the model with time and without the time. The statistical significance was measured using likelihood ratio test. If value of p is <0.05 after Benjamin–Hochberg correction for the number of tested shortest paths, the shortest path is considered to change significantly with time. Only significant shortest paths are kept.
2.3. Mediation analysis with PLS-SEM
Mediation analysis with PLS-SEM was performed to understand if the drug (APOM) had an effect on the disease state cells, that is, MPP+-treated SH-SY5Y cells. To this end, three latent variables (i.e., “Treat,” “Dis,” and “Med”) were built. Indicator genes for each latent variable were obtained from the united graph of total significant shortest paths on the KEGG pathway. Three groups of genes (i.e., 169 drug genes, 110 disease genes, and 948 mediator genes) found in shortest path analysis between drug-specific genes and disease-specific genes were used as indicator genes for each latent variable. That is, indicators of “Treat,” “Dis,” and “Med” were 169 drug genes, 110 disease genes, and 948 mediator genes, respectively. Not all genes in the set were used as indicator genes because the number of samples in the dataset was less than the number of genes in each set. For selecting indicator genes, a hierarchical clustering analysis based on correlation was performed.
After defining the latent variable, a mediation model was generated in which “Treat” was the independent variable and “Dis” was the dependent variable. The analysis allowed us to determine if the treatment had an effect on the disease and how it was determined if directly, by the mediator in both ways. Using PLS-SEM and the set of genes belonging to each latent variable, a mediation model was created and evaluated. These genes were considered as reflective indicators. This means that indicators are caused by latent variable. PLS models are characterized by two models: structural model and measurement model. The first one analyzes relationships between latent variables, whereas the second examines one relationships between observed (indicator) variables and latent variables. The structural model can be mathematically represented as follows:
where LV is the matrix representing the latent variables, B is the matrix of coefficients between latent variables, and E is the matrix of residuals. The measurement model for a particular latent variable n can be represented as follows:
where OVn is the block matrix of observed variables that generates latent variable LVn;
For model evaluation, two types of measures exist. The first ones are for evaluating the structure model [Eq. (1)]. The model was constituted by connections between latent variables. The second ones are for evaluating the goodness of the measurement model [Eq. (2)]. The model was constituted only by latent variables and their relationships with indicators. For structure model evaluation, R2 was used. It indicates how much variance of the endogenous latent variable is explained by independent latent variables. It is possible to classify the goodness of model based on the following criteria according to Sanchez (2013): (1) Low: R2 < 0.30 (for some authors <0.20); (2) Moderate: 0.30 < R2 < 0.60 (or 0.20 < R2 < 0.50); and (3) High: R2 > 0.60 (or >0.5). For measurement models, Dillon–Goldstein's rho and communality were used. The Dillon–Goldstein's rho explains to which extent the constructs can explain indicators. Its acceptable value should be >0.7. The communality corresponds to squared correlations between latent variables and its indicators. In other words, it measures shared variance between latent variables and their indicators. An acceptable value of communality is 0.5.
3. Results
3.1. Exploring disease and drug genes from microarray data
As SH-SY5Y cells exposed to MPP+ mimic PD, responsive genes to MPP+ treatment may be considered as disease-associated genes or simply disease genes. On the contrary, responsive genes to treatment of MPP+ with APOM might be regarded as drug-associated genes or simply drug genes because APOM is a nonselective dopamine agonist for PD. Therefore, disease or drug genes could be detected by microarray experiment that allows us to measure gene expression response or change at genomic scale. In this study, two time-series microarray data were used from our previous studies (Kim et al., 2013; Do, 2017) for the detection of disease genes and drug (APOM) genes in PD cell model. Both microarray data consisted of expression values measured at six time points (0, 3, 6, 9, 12, and 24 hours) from MPP+-treated SH-SY5Y cells and MPP+-treated SH-SY5Y cells with APOM for a total of 30,486 genes in the microarray. Genes with differential expression profile were identified with a Bioconductor package maSigPro (Conesa et al., 2006) known to support the analysis of time-series microarray data.
Disease genes were detected from the time-series microarray experiment data of MPP+-treated SH-SY5Y cells. The number of disease genes was 808, ∼2.7% of total genes on the microarray. Drug (APOM) genes were also detected from the time-series microarray experiment data of SH-SY5Y cells exposed to MPP+ with APOM. The number of drug genes was 935, ∼3.1% of total genes on the microarray. There were 175 common genes shared by disease genes and drug genes (Fig. 1), ∼11.2% of total response genes (disease genes+drug genes). These common genes can be interpreted as disease genes on which the drug (APOM) act directly or as disease genes on which the drug (APOM) does not have any effect.
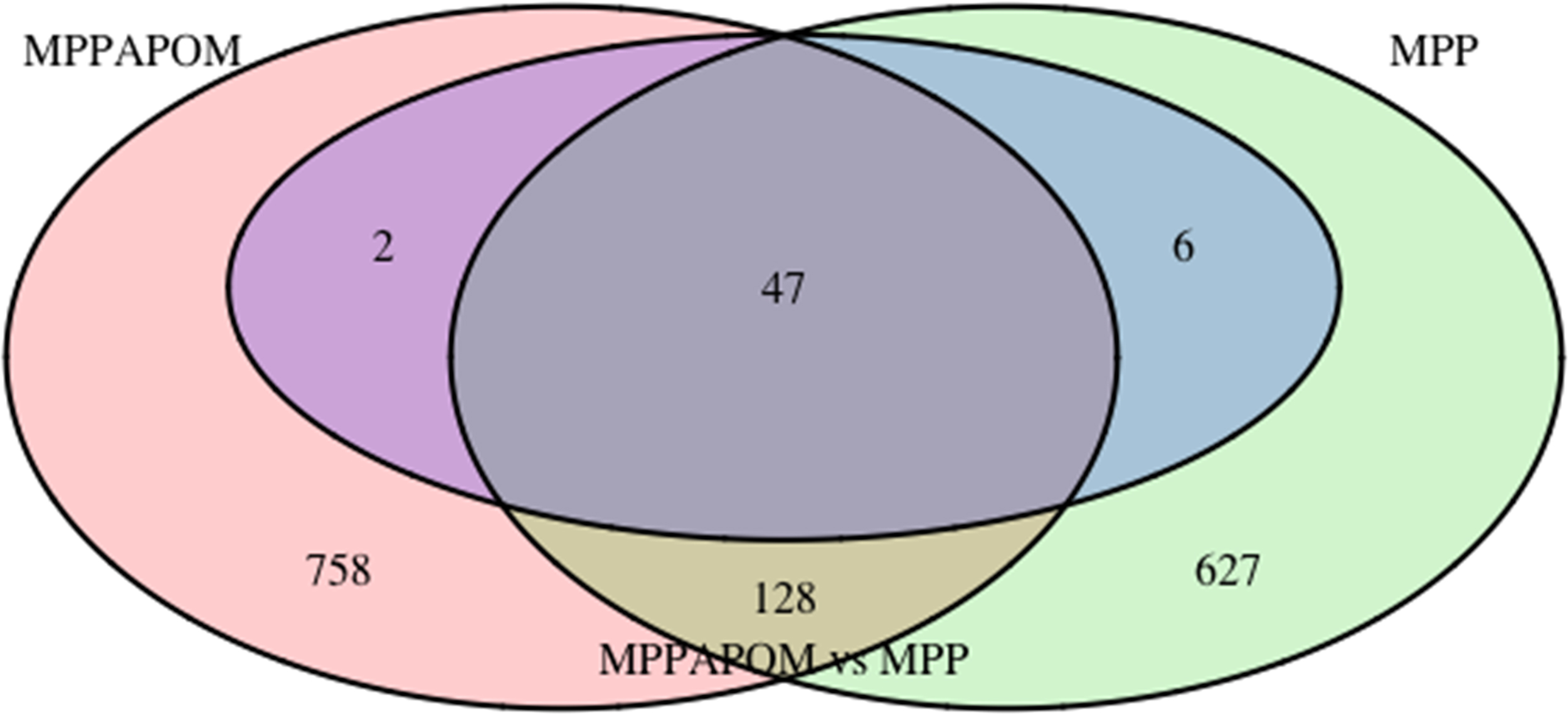
Venn diagram showing drug genes (MPPAPOM) and disease genes (MPP) detected from two groups of time-series microarray data.
To understand on which genes the drug (APOM) could act directly, a multi-series analysis was performed for the two time-series microarray data. Fifty-five genes showed differential profiles between the 2 groups of microarray data, of which 47 belong to the 175 common genes. Therefore, these 47 genes can be considered as disease genes on which the drug (APOM) can act directly. They are given in Table 1. Gene Ontology (GO) terms that were significantly overrepresented in these 47 genes were estimated with a web-based program Gorilla (Eden et al., 2009). Results are given in Table 2. GO terms for biological process, immune response-regulating cell surface receptor signaling pathway involved in phagocytosis (GO:0002433), positive regulation of peptidase activity (GO:0010952), and positive regulation of proteolysis (GO:0045826) showed significant enrichment scores with p-value <1.0E-3. Enriched GO terms for cellular component included external organelle (GO:0043230), membrane-bounded organelle (GO:0043227), and azurophil granule lumen (GO:0035578). This suggested that APOM in PD might directly target genes associated with autophagic and phagocytic pathways.
Forty-Seven Disease Genes Directly Affected by Apomorphine
Enriched Gene Ontology Terms Discovered by Gorilla Program in 47 Disease Genes Directly Affected by the Drug (APOMORPHINE) Versus Background Gene Set, Including All Genes on the Array
BP, biological process; CC, cellular component; GO, Gene Ontology.
3.2. Shortest path analysis between drug-specific and disease-specific genes
Among a total of 1568 response genes including disease and drug genes, there were only 175 common genes (Fig. 1). That is, the number of disease-specific genes was 633, whereas 760 genes were specific to the drug (APOM). Drug-specific genes might be indirectly associated with disease-specific genes. Indirect association between drug-specific and disease-specific genes could be understood by looking for connections between the two groups of genes. Using KEGG network obtained by merging all KEGG pathways (with R package graphite and KEGGgraph) and the shortest paths between drug-specific genes and disease-specific genes, it was possible to discover how the drug (APOM) effect propagated until disease genes were reached. The KEGG network included 5238 nodes and 73,070 connections. However, not all genes in the graph were present in the microarray experiment. For this reason, 77 genes were excluded from the graph. Therefore, analysis of shortest path model was performed on the KEGG graph constituted by 5161 genes and 71,636 connections. The number of direct shortest paths between drug-specific and disease-specific genes was 18,704.
To evaluate if the found shortest paths changed significantly with time for any gene that received a connection in the shortest path, an SEM analysis on each shortest path was performed (see Section 2.2 for details). The total number of significant shortest paths was 5263. Their union generated a graph of 1227 genes and 4602 connections. These 1227 genes of the graph can be distinguished into three groups of genes: (1) 169 drug genes from which the effect of drug starts, (2) 110 disease genes, and (3) 948 mediator genes that connect the drug and disease genes. To explore enriched pathways in each group of genes, R package clusterProfiler (Yu et al., 2012) was used. Figures 2 and 3 show significantly enriched KEGG pathways in 169 drug genes and 948 mediator genes, respectively. No KEGG pathway was enriched for 110 disease genes. Enriched pathways for 169 drug genes included carbon/amino sugar and nucleotide sugar/inositol phosphate metabolism, protein processing in endoplasmic reticulum, estrogen/cAMP/HIF-1/glucagon signaling pathways, and so on. In the case of 948 mediator genes, there are many enriched signaling pathways, including ras/dopaminergic synapse/oxytocin/insulin/apelin/sphingolipid signaling pathways. This indicates that drug genes might regulate disease genes through mediator genes that are related to many signaling pathways.

KEGG pathway enrichment analysis for 169 drug genes on the united graph of significant shortest paths. KEGG, Kyoto Encyclopedia of Genes and Genomes.
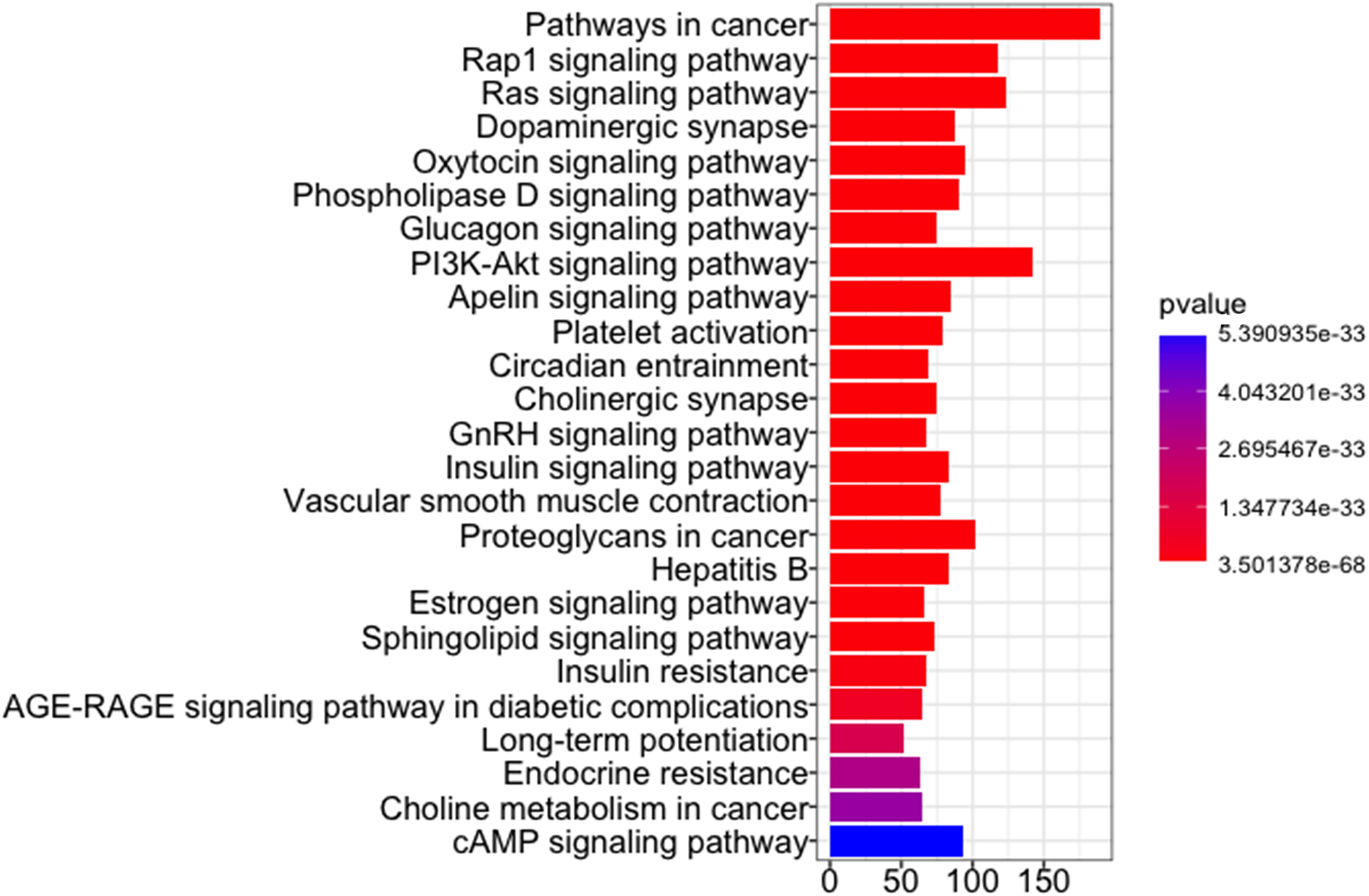
KEGG enrichment analysis for 948 mediator genes on the united graph of significant shortest paths.
3.3. Mediation analysis of mediator genes with PLS-SEM
In the previous section, three groups of genes were found through shortest path analysis between drug-specific genes and disease-specific genes (i.e., 169 drug genes, 110 disease genes, and 948 mediator genes). By using these three groups of genes, mediation analysis with PLS-SEM was performed to understand if drug genes have an effect on disease genes. To this end, 3 latent variables of “Treat,” “Dis,” and “Med” were built with 169 drug genes, 110 disease genes, and 948 mediator genes as their reflective indicators, respectively. Not all genes in the set were used because the number of samples in the dataset was less than the number of genes in each set. For selecting genes, a hierarchical clustering analysis based on correlation was performed. In the next step, PLS-SEM model was created and fitted. PLS-SEM is characterized by two models: structural model and measurement model. The first one analyzes relationships between latent variables, whereas the second one examines relationships between indicator variables (here, genes) and the latent variables. Therefore, feasibility measure of PLS-SEM is separately evaluated for structural model and measurement model.
The feasibility of measurement model was evaluated by Dillon–Goldstein rho value to explain to which extent latent variables could explain indicator variables. An acceptable value should be >0.7. In our case, Dillon–Goldstein rho values for the measurement model were 0.98, 0.95, and 0.99 for latent variables of “Treat,” “Dis,” and “Med,” respectively. Another important feasibility measure is communality, which corresponds to squared correlations between latent variables and its indicators. In other words, it measured shared variance between latent variables and their indicators. An acceptable value of communality is 0.5. In our case, communality values were 0.85, 0.90, and 0.85 for latent variables of “Treat,” “Dis,” and “Med,” respectively. For structure model evaluation, we used R2 to show how much variance of the endogenous latent variable was explained by independent latent variables. In our model, we had only an independent latent variable, “Treat.” Mean R2 was 0.52, a moderate value of R2.
When analyzing the model, it is important to consider estimated value of path coefficient that represents the response of dependent variable to a unit change in an independent variable when other variables in the model are fixed. The path coefficient can be interpreted as the connection strength or effect strength between latent variables. The significance of path coefficient was evaluated with a bootstrap approach. As given in Figure 4, all path coefficients are significant if 95% confidence intervals do not include zero. The direct effect from latent variable “Treat” to “Dis” was −0.26. The indirect effect through “Med” was −0.35 (by multiplying path coefficients involved in the indirect path, i.e., −0.45 × 0.77). Total effect (direct effect + indirect effect) was −0.61. This means that, if we increase global expression of drug genes, they can reduce global expression of disease genes by direct and indirect ways. This indicates that drug genes responding to APOM might contribute to negative regulation of disease genes by direct or indirect ways.
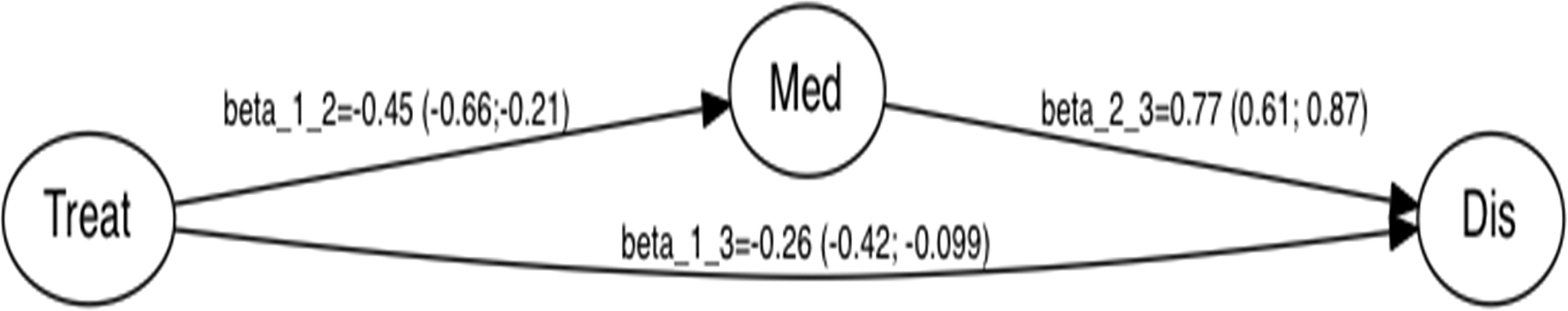
Path diagram for structure model showing relationships among three latent variables. The number on arrow represents path coefficient with 95% confidence interval shown inside parenthesis.
4. Discussion and Conclusions
Unlike levodopa that requires conversion into dopamine to be effective for PD, APOM directly plays a role as a nonselective dopamine agonist. APOM is used mainly for advanced PD patients showing no response to levodopa or other agonists (Okun and Foote, 2010). APOM has functions as a potent antioxidant and a free radical scavenger besides its receptor-mediated action (Gassen et al., 1996). However, it has been reported that long-term use of APOM in PD treatment brings about side effects such as nausea, orthostatic hypotension, and fibrotic nodules at the site of APOM injection (Deleu et al., 2004). For safe use of APOM in PD treatment, it is essential to understand the molecular mechanisms involved in its effect.
In this study, we examined mediated effects of APOM in PD cell model by using two groups of time-series microarray data. Disease genes and drug genes were identified from microarray data of MPP+-treated SH-SY5Y cells and APOM-added MPP+-treated SH-SY5Y cells, respectively. Numbers of disease genes and drug genes were 808 and 935, respectively. The number of common genes between disease and drug genes was 175, ∼11.2% of total response genes (disease genes and drug genes). Among these 175 common genes, expression profiles of 47 genes showed significant changes by the drug (APOM). Therefore, these 47 genes could be considered as disease genes on which APOM can act directly. Enriched GO terms in these 47 genes were GO:0002433 (immune response-regulating cell surface receptor signaling pathway involved in phagocytosis), GO:0002431 (Fc receptor-mediated stimulatory signaling pathway), GO:0045862 (positive regulation of proteolysis), and so on (Table 2).
Drug-specific and disease-specific genes were obtained after excluding the 175 common genes in their gene group, respectively. Drug-specific and disease-specific genes might be linked by mediator genes. On the graph obtained by merging all KEGG pathways, shortest paths linking drug-specific genes and disease-specific genes were evaluated with SEM. The number of genes on the graph generated by merging all significant shorted paths was 1,227. These 1,227 genes can be distinguished into 3 groups: 169 drug genes, 110 disease genes, and 948 mediator genes. In the pathway enrichment analysis for 948 mediator genes, many signaling pathways including ras/dopaminergic synapse/oxytocin/insulin/apelin/sphingolipid signaling pathways showed enrichment. This suggests that drug genes might regulate disease genes through mediator genes that are related to signaling pathways.
To explore the role of mediator genes toward disease genes, mediation analysis with PLS-SEM was performed by using expression values of three group of genes (i.e., 169 drug genes, 110 disease genes, and 948 mediator genes). Three latent variables of “Treat,” “Dis,” and “Med” were built with 169 drug genes, 110 disease genes, and 948 mediator genes as their reflective indicators, respectively. The direct effect from latent variable “Treat” to “Dis” was −0.26. The indirect effect through “Med” was −0.35 and the total effect was −0.61 (Fig. 4). This indicates that drug genes responding to APOM might contribute to negative regulation of disease genes by direct or indirect ways. Our result supports that APOM has effectiveness as drug on PD cell model in terms of gene expression regulation.
Footnotes
Author Disclosure Statement
The authors declare they have no competing financial interests.
Funding Information
This study was supported by a grant (No. 2012R1A1A2005622) of the Basic Science Research Program through the National Research Foundation (NRF) of Korea funded by the Ministry of Education, Science, and Technology (MEST).