
Abstract
Evolutionary models of proteins are widely used for statistical sequence alignment and inference of homology and phylogeny. However, the vast majority of these models rely on an unrealistic assumption of independent evolution between sites. Here we focus on the related problem of protein structure alignment, a classic tool of computational biology that is widely used to identify structural and functional similarity and to infer homology among proteins. A site-independent statistical model for protein structural evolution has previously been introduced and shown to significantly improve alignments and phylogenetic inferences compared with approaches that utilize only amino acid sequence information. Here we extend this model to account for correlated evolutionary drift among neighboring amino acid positions. The result is a spatiotemporal model of protein structure evolution, described by a multivariate diffusion process convolved with a spatial birth–death process. This extended site-dependent model (SDM) comes with little additional computational cost or analytical complexity compared with the site-independent model (SIM). We demonstrate that this SDM yields a significant reduction of bias in estimated evolutionary distances and helps further improve phylogenetic tree reconstruction. We also develop a simple model of site-dependent sequence evolution, which we use to demonstrate the bias resulting from the application of standard site-independent sequence evolution models.
1. Introduction
Protein alignment is an integral part of bioinformatic analyses and is a classic widely studied problem in computational biology. Existing methods for aligning two or more proteins compare amino acid sequences and/or structures of the proteins, and encompass a variety of algorithms with different strengths and purposes. Such algorithms are a fundamental part of phylogenetic research in particular, where the degree and nature of evolutionary divergence between species is a quantity of interest. Alignment procedures that are widely used in studies of protein evolution are based only on the amino acid sequence and do not incorporate the tertiary (three-dimensional) structure of the proteins. Methods that do incorporate tertiary structure, such as those mentioned in Wang et al. (2013), do not account for the evolution over time of those structures.
Recently, Challis and Schmidler (2012) introduced a stochastic evolutionary model of protein sequence and structure for this purpose; however, their approach, similar to the vast majority of alignment algorithms, assumes that “sites” (individual amino acid characters or backbone atom coordinate triples) evolve independently of one other. This assumption is well known to be violated since amino acid identities and spatial locations are highly dependent due to a combination of physicochemical constraints and interactions, including bond lengths and excluded volume, hydrophobic and electrostatic attraction and repulsion, hydrogen bonding, and other cooperative effects in forming stable local and global protein structure. Nevertheless, alignment algorithms based on both sequence and structural information typically ignore the correlations induced by these interactions. Ignoring dependence is often justified by the computational intractability of site-dependent models (SDMs) (Challis and Schmidler, 2012; Herman et al., 2014). In this article we demonstrate that in structure-based alignment, as in sequence-based, ignoring site dependence systematically biases evolutionary inference. We present an expanded version of the Challis and Schmidler model that incorporates neighbor dependence without sacrificing computational tractability.
1.1. Motivation
von Haeseler and Schöniger (1998) examined the effect of site dependence on estimates of evolutionary distance between pairs of biological sequences. Using a model of whale mitochondrial DNA evolution whereby the sequence evolves as a collection of independent subsequences, each exhibiting Markovian dependence among its amino acids, the authors demonstrated the tendency to underestimate the true evolutionary distance between two sequences when using a site-independent model (SIM). Figure 3a replicates this effect using binary sequences from a nearest-neighbor site-dependent sequence model that does not assume independent subsequences, described in Section 5. When estimating the divergence time for these sequences under a site-independent version of the same model (
Despite a variety of efforts, no site-dependent sequence model has emerged as a widely applicable replacement for commonly used site-independent sequence models (Arenas, 2015). The primary hurdle to doing so is computational—adding realistic dependence generally prohibits the use of efficient alignment algorithms that rely on dynamic programming. We address this issue further in Section 5.
In contrast, we demonstrate in Section 3 that the site-independent structural model of Challis and Schmidler (2012) can be extended to a site-dependent structural model, incorporating site dependence while maintaining the same interpretability and mathematical and computational tractability as the SIM. Thus, we can incorporate dependence into the evolutionary structural part of the model in a relatively straightforward way. Using data simulated from the SDM, we find a systematic underestimation effect for structural data due to the independent-site assumption, similar to that observed in sequences (Fig. 3e). The new SDM can then be paired with a sequence evolution model to provide a site-dependent expansion of the joint sequence-structure model of Challis and Schmidler (2012).
The article is organized as follows. We briefly review the SIMs commonly used for sequence evolution as well as the structural diffusion model of Challis and Schmidler (2012). We then describe the general form of a dependent structural diffusion model. Section 3 describes the details of incorporating dependence into the model, with computational tractability being the key constraint on the model's form. Section 4 describes a reparameterization of the SDM necessary for analyzing the SDM's effect on phylogenetic inference. Section 6 revisits the motivating aforementioned example and compares inferences and phylogenies from the expanded model on a number of real protein examples. Section 5 describes the basic site-dependent sequence model used in this article.
2. Site-Independent Models (SIMS) of Protein Evolution
We briefly review the site independence assumption as applied in familiar sequence evolution models, as well as more recent developments in modeling structural evolution.
2.1. Sequence evolution
Stochastic models of protein sequence evolution are widely used for statistical alignment, evolutionary inference, and phylogenetic reconstruction (Challis and Schmidler, 2012; Herman et al., 2014; Wang and Schmidler, 2014). The general form taken for such models is similar to that used in Challis and Schmidler (2012), where the joint likelihood for the two sequences
where
2.2. Challis–Schmidler model
Challis and Schmidler (2012) introduced a stochastic model for protein structure evolution, extending a previously developed probabilistic framework for structural alignment of proteins (Schmidler, 2006; Wang and Schmidler, 2014) into a model suitable for the study of molecular evolution. This study demonstrated the ability to significantly improve phylogenetic inference when structural information about the proteins is available (see also Herman et al., 2014). We briefly review the original Challis–Schmidler model here, before introducing our extended model incorporating site dependence in the next section. Throughout the article, these structural models will be referred to as the SIM and SDM, respectively.
The diffusion of individual
where
We refer the reader to Challis and Schmidler (2012) for a more detailed discussion of the diffusion process described earlier and for various other model details omitted here for brevity. Finally, we give the likelihood function resulting from the aforementioned structural model. Let
2.3. Bayesian inference
The models described earlier provide the likelihood of the data which can be used to estimate alignments, evolutionary trees and distances, and model parameters. Throughout this article we will follow the Bayesian paradigm, under which inference is based on the posterior distribution:
where the likelihood
3. A Site-Dependent Structural Diffusion Model
We now proceed to extend the aforementioned SIM of protein structure evolution to incorporate site dependence. As will be shown, computational tractability can be preserved in the case where dependence is limited to nearest-neighbor relationships.
3.1. Dependence in a multivariate OU process
The independent site model (Eq. 2) can be written as a multivariate diffusion in the form
where
For purposes of this article we set
Mean Sample Correlations Between Dimensions and Mean lag-1 Autocorrelations Along Dimensions for 71 Globin and 478 MALIDUP Protein Structures
Under the SDM then, the joint evolution of the
with

where
where the stationary covariance

Although these closed-form solutions exist for general
3.2. Computational tractability in phylogenetic models
Common uses of evolutionary models, in phylogenetic or homology detection contexts, require the ability to optimize or average over the set of possible alignments. In a Bayesian or maximum likelihood context, the alignment must be inferred simultaneously with the other parameters. Because of the (exponentially large) size of the alignment space, algorithmic efficiency considerations in these calculations play a key role. In particular, calculating the joint likelihood
with X and Y denoting ancestor and descendant structures, respectively. Models with long-range dependence among sites, including the dependent diffusion model (Eq. 5) with general
3.3. Constructing a dependent structural diffusion model
A natural approach to introducing limited neighbor dependence into the diffusion model is to consider the backbone sites' coordinates as a series of nodes with forces acting upon each pair of neighboring sites, for example, as in a ball and spring model. Figure 1 shows a general ball and spring model with spring constants

General ball and spring model for n backbone positions.
The corresponding Gaussian model with neighbor dependence is a spatial first-order autoregressive process, denoted AR(1). However, setting the spring matrix equal to an AR(1) precision matrix gives a set of equations for the spring constants
We used symbolic algebra software to assist in solving for general matrices
where
Similar computer algebra experiments were used to demonstrate that no such solutions exist for any diffusion of the form in Eq. (5), where
3.4. Bayesian inference for the SDM
Under the new SDM specified by Eqs. (5) and (11), the joint distribution
where
Bayesian inference based on this joint distribution (and that including indels) uses priors and sampling techniques detailed in Challis and Schmidler (2012) with trivial additions to accommodate priors and sampling for the model's dependence parameter
3.5. Modified dynamic programming for pair HMMs with nearest-neighbor dependence
The recursive equations used for the pair HMM underlying the SIM (Durbin et al., 1998) require several modifications to be used with the SDM. These modifications are specific to the form of
In the SDM, the dynamic programming equations' coordinate emission probabilities for each site will now involve preceding positions' coordinates. Because these probabilities are specified by distributions conditional on an alignment, we must know the form of the joint distribution
In our model, as in Challis and Schmidler (2012), a pair HMM is used to model the distribution of pairwise alignments between two proteins. As described in Durbin et al. (1998), the use of a pair HMM allows one to calculate the probability of two protein structures marginalized over all possible alignments between the two structures. This is accomplished through dynamic programming by using the well-known forward algorithm to recursively calculate values of

where
To illustrate the set of changes needed, we focus only on the Match equation (Eq. (13)); analogous changes are required for the other two recursive equations. Equation (13) gives the total probability of all alignments up to position
where the superscripts on
To highlight an important point regarding Eq. (16), note that the emission distribution for a matched pair given a previous Match (
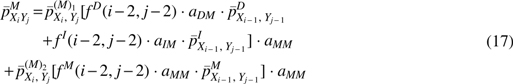
The presence of the recursive term
3.5.1. Derivation of emission probabilities
Suppose
where
with
The joint distribution
4. Joint Sequence-Structure Model for Phylogenetic Inference
Phylogenetic inference involves constructing a phylogenetic tree using estimates of the evolutionary distance between proteins, or equivalently models of the time-dependent evolution. Traditionally this is done using site-independent sequence evolution models parameterized by a matrix Q of relative substitution rates, defining a likelihood over the time
4.1. Amino acid sequence model
The sequence portion of our joint sequence and structure model follows that used in Challis and Schmidler (2012), given in Eq. (1).
4.2. Site-dependent random effect model
In a sequence evolution model (Eq. 1), only the product
However, this means that disagreement between the structural evolution model and sequence evolution model regarding the divergence time
The importance of distinguishing these two quantities is highlighted by the plot in Figure 2, where we estimated divergence time separately using the sequence-only model of Eq. (1) and the independent structure-only model (see e.g., Challis and Schmidler, 2012) for a set of globins. There is a strong arguably linear relationship between the structure-only evolutionary distance
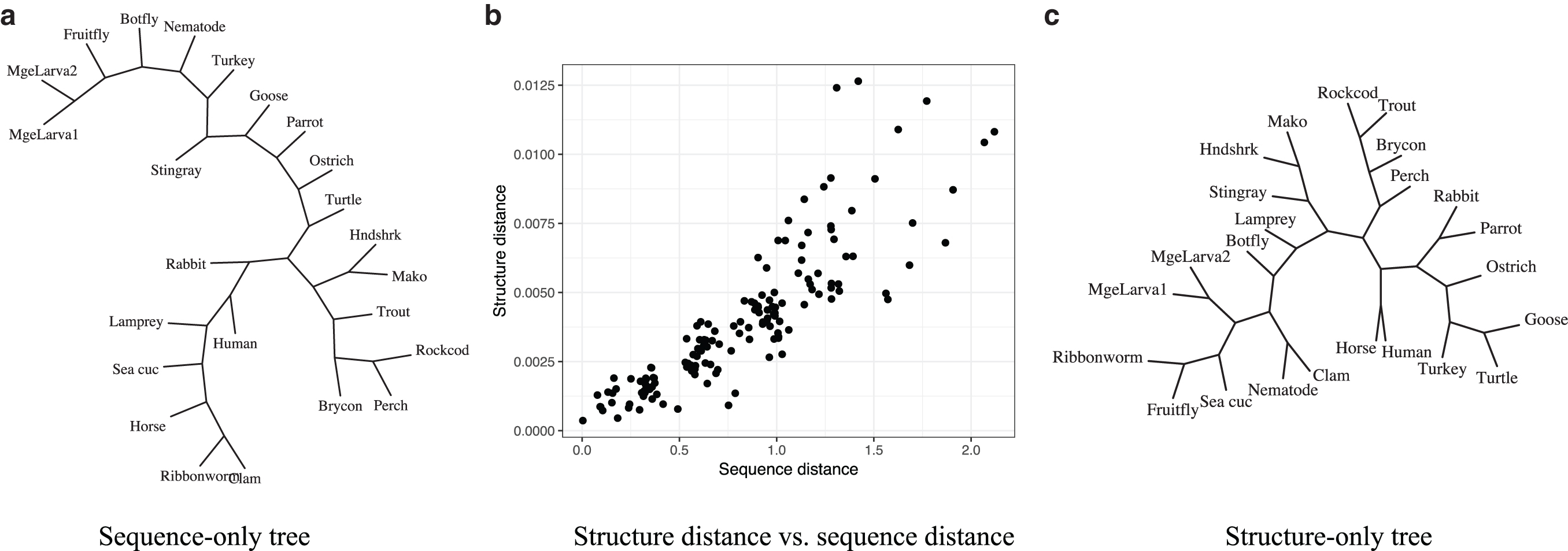
Pairwise sequence-only distance (
Instead, we introduce a random effect model defining a stochastic linear relationship between sequence and structure distances:
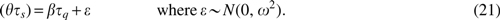
Here
and the PDE governing the structural diffusion is

To ensure the structure distance variable
5. An SDM for Binary Sequence Evolution
As mentioned earlier, the vast majority of applied work on statistical alignment and phylogenetic inference utilizes SIMs. Here we introduce a simple SDM of sequence evolution, analogous to the SDM of structure introduced in Section 3.3, to explore the impact of the SIM assumption when the data actually exhibit site dependence. Although we restrict our model to a simple case (binary sequences), it yields insights into the effect of mis-specifying site independence that provide a cautionary note for the widespread use of these models. Extension to more realistic SDMs for sequence is of significant interest, and would allow corrections similar to those exhibited for the structural SDM in Section 6.
5.1. Model
Let x or y represent a length n binary sequence. The space of all
To construct a simple model for site-dependent evolution of x to y, we introduce a Markov chain on
with
This model is a reparameterization of one-dimensional Ising model, a well-known model for ferromagnetism in statistical mechanics, where b is the usual Ising dependence parameter. The stationary distribution can be written as
where Z is the normalizing constant
Suppose the Markov chain is currently in state i. After an exponential waiting time elapses (given by a sample from the distribution
Given b, data can be simulated from this model for modest n as follows. First, the transition probability matrix
This simulation scheme is practical only for modest n because of the computational difficulty in exponentiating a large n-by-n matrix. For the binary sequence simulations used in Figure 3, we simulated subsequences of length 8. For a more detailed treatment of a similar site-dependent model based on subsequences, see von Haeseler and Schöniger (1998).
6. Results
All inferences were performed on the Duke Computer Cluster (DCC), a heterogeneous network of shared computing nodes; a typical node CPU is an Intel Xeon 2.6 GHz. Average runtimes for the SIM range from 20 to 60 iterations per second depending primarily on the length of the proteins, whereas SDM computations are roughly an order of magnitude slower than the SIM. All model parameters were sampled via random walk Metropolis-Hastings, augmented with a library sampling step for rotation parameter R as described in Challis and Schmidler (2012).
6.1. Improved estimation of evolutionary distances
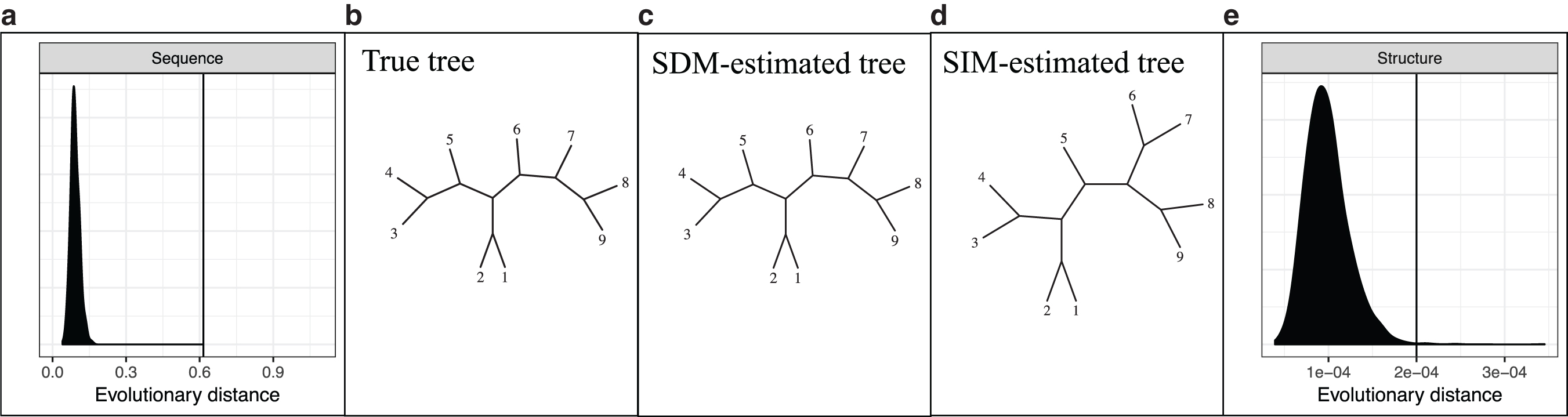
The posterior distribution shown in Figure 3a demonstrates the tendency of site-independent models to underestimate the true evolutionary distance between two sequences when the sequences arise from a process with site dependence (in this case, the correlated-neighbor model described in Section 5). The SIM significantly underestimates the true value, reflecting the significant bias in the SIM's MLE. This is likely a conservative estimate of the bias compared with that for real sequences, which typically exhibit some long-range dependence not present in our dependent binary sequence model.
Figure 4 similarly shows posterior distributions obtained under the (structural) SIM when applied to structures exhibiting neighbor-dependent evolution. The left panel of Figure 4 shows the posteriors from both the SIM and SDM. We see again that the SIM underestimates the true evolutionary distance, whereas the SDM corrects for this.
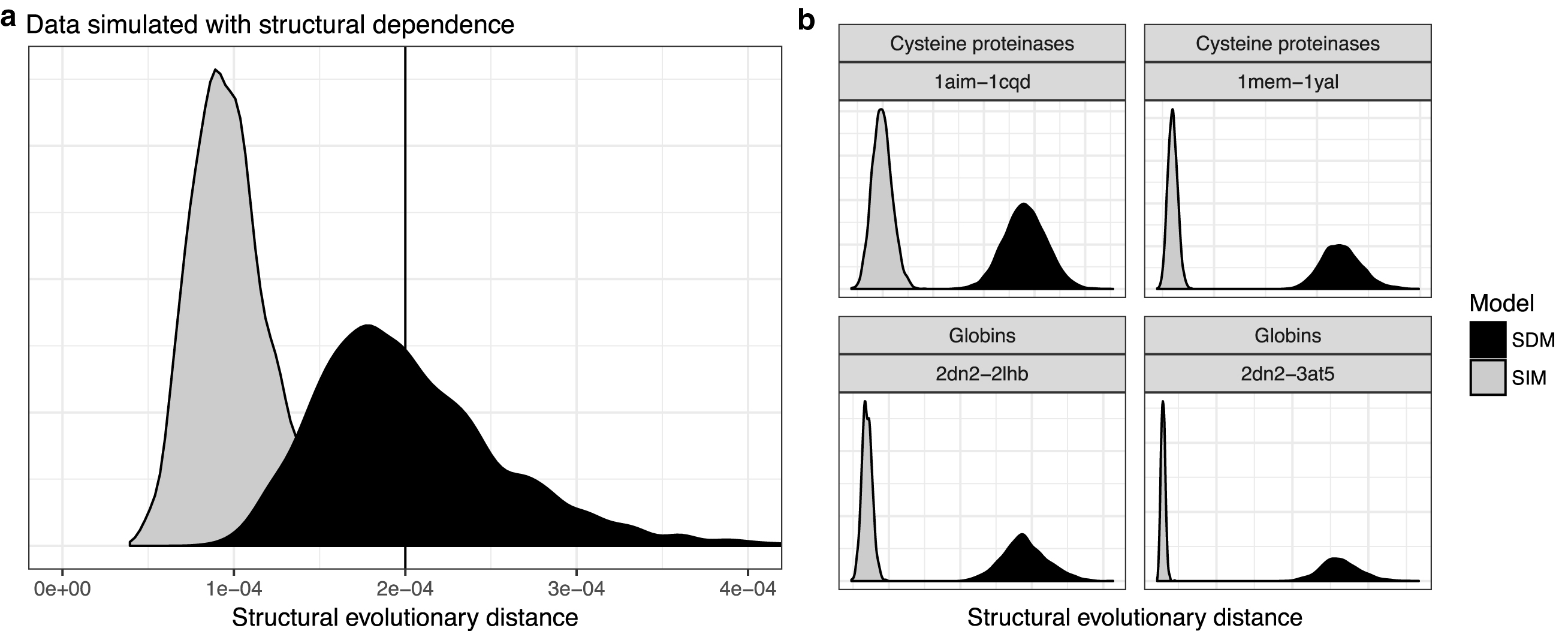
Estimation of evolutionary distance using SIM (light) and SDM (dark), for
Although this is not surprising on data simulated from the SDM, similar results are observed on real data for which the “true” distance is unknown. The four plots at right in Figure 4 compare the SIM and SDM posterior distributions for structural distance
6.1.1. Non-neighbor dependence
Proteins exhibit significant non-neighbor dependencies due to shared environments and physicochemical interactions between amino acids that are distant in sequence but proximal in space. Simulations were run using general (nonbanded) covariance matrices to simulate structural evolution with long-range correlations, with the SDM then used to estimate evolutionary distance. The results (omitted for brevity) are very similar to the left panel of Figure 4: the SIM noticeably underestimates the true structural distance, whereas the SDM accurately estimates it. This indicates the robustness of the nearest-neighbor approximation, required for efficient computation, to more general dependency patterns.
6.2. Effect on phylogeny of ignoring structural dependence in globin structures
Errors in estimation of pairwise evolutionary distances have the potential to undermine phylogenetic inference as well. To explore this, we compare phylogenetic trees reconstructed via neighbor-joining for a group of 16 globins using the SIM versus that obtained under the SDMre of Section 4. In each case, the respective model was used to estimate the pairwise distances for all pairs of proteins, and the resulting pairwise distance matrix was used to produce a neighbor-joining tree with the PHYLIP and Drawtree software (Felsenstein, 1989). Differences observed in these trees can be expected to also appear in trees if the SDM were used to replace the SIM component of the fully Bayesian joint sequence-structure tree estimation (Herman et al., 2014).
The phylogenetic trees estimated using posterior mean evolutionary distances are shown in Figure 5. The SIM and SDMre trees are very similar, and neither matches the accepted NCBI taxonomy exactly. However, the SDMre tree improves upon the SIM tree in that botfly and fruit fly are now placed together in a single clade with no other species, as in the NCBI taxonomy. This example demonstrates that phylogeny estimation can be adversely affected by ignoring structural dependence, even for proteins with high structure similarity such as these globins.
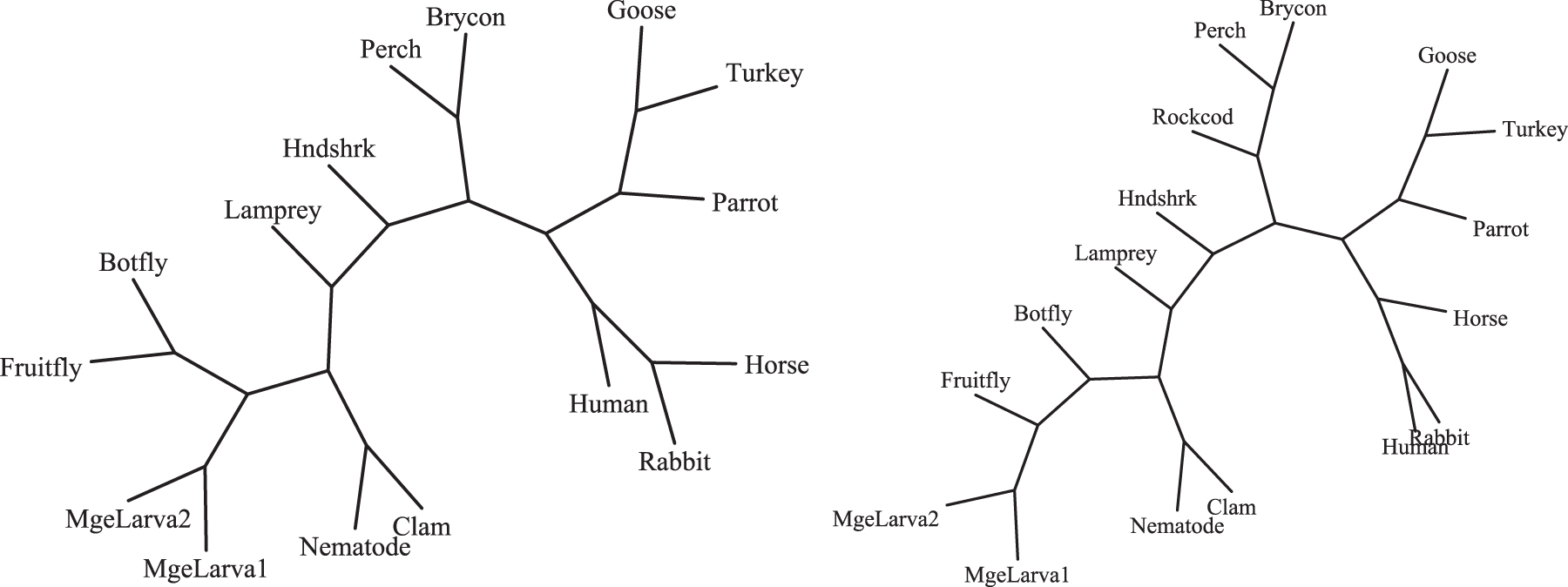
The SDMre tree (left) improves upon the SIM tree (right) by grouping the botfly and fruit fly in their own clade, matching the accepted NCBI taxonomy.
The SIM and SDMre models leading to the trees in Figure 5 differ in two ways: incorporation of dependence in the diffusion, and incorporation of the random effect relation between the sequence and structure time parameters. For comparison, we also ran the SIM with the random effect incorporated, but without dependence in the diffusion model. This SIMre does not correctly group botfly and fruit fly, indicating that it is the site dependence that leads to the improved tree topology. For comparison, the sequence-only tree is also shown (for a superset of globins) in panel (a) of Figure 2; it is highly inaccurate due to many pairs with highly divergent sequences. Without the structural component of the model included, these divergent sequences yield highly uncertain distance estimates that significantly destabilize the tree.
7. Discussion
The site-dependent structural evolution model described here allows a significant improvement in model realism while retaining the computational tractability necessary for use in phylogenetic inference. As shown, the incorporation of dependence into the model significantly reduces bias in the estimates of evolutionary distance, and can have a resulting stabilizing effect on phylogenetic tree reconstruction. These results suggest a need for continued research on computationally efficient site-dependent sequence evolution models, which can be expected to further improve inference in these problems. This is because our current combined sequence-structure model pairs the site-dependent structural model with a site-independent sequence model, which likely still retains some downward bias on the estimated evolutionary distance due to the independence assumption in the sequence side of the model.
A natural next step will be to incorporate the site-dependent structural model presented here into the fully Bayesian simultaneous alignment and phylogeny reconstruction model of Herman et al. (2014), which currently uses the site-independent structural model. This extension would be straightforward and may improve inference of multiple sequence alignments in addition to improving inference of phylogenetic trees.
8. Software
The code base for this article is written in R and C
Footnotes
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This study was partially supported by NSF grant DMS-1407622 and NIH grant R01-GM090201 (S.C.S.). J.L.T. was supported by NIH grant GM118508. G.L. was partially supported by NSF training grant DMS-1045153 (S.C.S.).