
Abstract
Methods previously developed by the author are applied to uncover several sites of interest in the spike glycoproteins of all known human coronaviruses (hCoVs), including SARS-CoV-2 that causes COVID-19. The sites comprise three-dimensional neighborhoods of peptides characterized by four key properties: (1) they pinpoint regions of high free energy in the backbone whose obstruction might interrupt function; (2) by their very definition, they occur rarely in the universe of all gene-encoded proteins that could obviate host response to compounds designed for their interference; (3) they are common to all known hCoV spikes, possibly retaining activity in light of inevitable viral mutation; and (4) they are exposed in the molecular surface of the glycoprotein. These peptides in SARS-CoV-2 are given by the triples of residues (131, 117, 134), (203, 227, 228), and (1058, 730, 731) in its spike.
1. Introduction
At this moment, it is hardly necessary to pontificate about the pressing need for an effective and robust vaccine to treat COVID-19 for human society and humanity itself are under siege. Tools developed over the past year and a half, recently announced by Mary Ann Liebert, Inc. (2020) and published in Penner (2020), have already been shown to be an effective predictor for the reconformation of viral glycoproteins in vitro. The input to the method is the three-dimensional (3D) structure of a viral or other protein in the form of a Protein Data Bank (PDB) file (Berman et al., 2000). Viral adsorption and fusion, two necessary steps in infection, are well known (cf. Levine, 1992; Dimmock et al., 2007) to require such reconformation, which depends upon regions of the glycoprotein backbone with high free energy for their actuation according to Penner (2020). Specific residues targeting conformational change, which moreover persist across the spike glycoproteins of all known human coronaviruses (hCoVs), are computed here and proposed as sites of interest.
In fact, obstruction to reconformation by blocking these reservoirs of high free energy could interrupt spike function and thereby block infection. Determination of these reservoirs is accomplished using a standard tool of protein theory, the Pohl-Finkelstein quasi-Boltzmann Ansatz, observed by Pohl (1971) and explained by Finkelstein et al. (1995a,b) and Finkelstein and Ptitsyn (2016), applied to a database of protein backbone geometry computed in Penner et al. (2014). By their very definition, high free energy regions correspond to protein backbone geometries that are rare in the universe of all proteins and shall here sometimes be termed exotic. These exotic regions are thus likely to be rare in the host organism as well, hence their targeted interference might be relatively benign.
Another consideration for RNA viruses such as CoVs, which typically have high rates of mutation and antigenic drift according to Elena and Sanjuán (2005), is that a favorable target should be resilient against such variations. The approach here to this aspect is to propose sites of interest with the previously discussed attributes that furthermore are common in the sense of surmised functional alignment across all hCoV spikes, with the presumption that this commonality would likewise be shared by eventual different strains or more extensive mutations in future.
As a practical matter, any such salutary site must also lie in an exposed glycoprotein surface region and not be buried in its interior, in order that attachment or obstruction might be sterically feasible. It is fortunate that for the SARS-CoV-2 spike, there actually are sites satisfying all these requirements, namely 3D neighborhoods of residues 131, 203, and 1058.
2. Background
Various facts are first recounted to set the stage.
2.1. Human coronaviruses
There are at least seven CoVs afflicting humans (hCoVs) (cf. Pyrc et al., 2007) (with the corresponding disease names given in parentheses): SARS-CoV-1 (SARS), MERS-CoV (MERS), SARS-CoV-2 (COVID-19, or simply COVID), and the endemic hCoV diseases NL63, HKU1, OC43, and 229E, whose corresponding viruses are indicated with the suffix -CoV as for MERS. For each of these hCoVs and for CoVs in general, adsorption and fusion are affected by spike glycoproteins.
The spikes for SARS-CoV-1, SARS-CoV-2, and NL63-CoV all bind to Angiotensin-converting enzyme 2 (ACE2) by Li et al. (2003), Walls et al. (2020), Wu et al. (2009), MERS-CoV to Dipeptidyl Peptidase 4 (DPP4) by Wang et al. (2013), HKU1-CoV and OC43-CoV to 9-O-acetylated sialic acids by Hulswit et al. (2019), and 229E-CoV to Aminopeptidase N (APN) by Bonavia et al. (2003). As determined by Clustal Omega explained in Smith et al. (2011), the spikes for SARS-CoV-1 and SARS-CoV-2 have high homology identity (76%) but relatively weak homology with NL63-CoV (30% and 31%, respectively). NL63-CoV and 229E-CoV spikes have surprisingly high homology identity (63%), with all other pair-wise identities of these seven in the 30%–40% range. MERS, SARS, and several endemic hCoV diseases appear to confer short- to medium-term immunity against reinfection according to Lipsitch (2020) and Aldridge et al. (2020). The endemic hCoV diseases are seasonal, favoring December and January, according to Gaunt et al. (2010), whereas MERS proliferates mostly in June as claimed by Nassar et al. (2018). COVID seasonality and reinfection immunity remain pressing open questions.
2.2. The spike glycoprotein
The CoV viral envelope is on the order of 80–100 nm diameter, with each spike glycoprotein standing at a height of ∼15–20 nm above it. Like the hell-dog Cerberus, at the virus envelope-distal end of the trimer spike lie three heads, each with its own receptor binding domain, which can presumably independently oscillate between an up (or standing/open) and a down (or lying/closed) conformation, and owing to steric constraints, it is only in the up configuration that ACE2 binding can occur for SARS-CoV-1 (Kirchdoerfer et al., 2018). For hCoVs in general, there are another three domains of the spike, to be called lobes, nearby the heads when up and contiguous when down, which occur in the following pattern: the head in chain A (B and C, respectively, in this counter-clockwise order when viewed from beyond the virus envelope-distal end) is proximal to the lobe in chain B (C and A). In examples for MERS-CoV and SARS-CoV-1 that are wild-antibody-bound as described in Walls et al. (2019b); Pallesen et al. (2017); and Wang et al. (2019), the epitopes also lie on the heads.
The presumptive fusion peptide from SARS-CoV-1 in Walls et al. (2019a) lies centrally located between the three heads and is completely obstructed when the heads are down. Upon binding, the spike undergoes dramatic reconformation, according to Walls et al. (2017), the details of which are not known, although finally providing the 6-helix bundle characteristic of a Class I fusion protein, as explained in White et al. (2008) and Bosch et al. (2003). Many of the hCoVs including SARS-CoV-2 are known to enter the cell through endocystotis (cf. Wang et al., 2008; Burkard et al., 2014; Ou et al., 2020). In several investigated examples, the spike is covered with an elaborate glycan shield (as in Ströh and Stehle, 2014; Walls et al., 2016; Vandakari and Wilce, 2020). Both MERS-CoV and SARS-CoV-2, but not SARS-CoV-1, support a furin cleavage site that may enhance fusion, according to Walls et al. (2020); Millet and Whittaker (2014); and Belouzard et al. (2009).
2.3. hCoV spike PDB files
There are 45 PDB files for spike glycoproteins of hCoVs: 3 for SARS-CoV-2 given in Walls et al. (2020) and Wrapp et al. (2020); 18 for SARS-CoV-1 in Kirchdoerfer et al. (2018), Gui et al. (2017), Yuan et al. (2017), Song et al. (2018), and Walls et al. (2019b); 19 for MERS-CoV in Song et al. (2018), Walls et al. (2019b), Pallesen et al. (2017), Wang et al. (2019), and Park et al. (2019); 2 for OC43-CoV in Tortici et al. (2019); and 1 each for 229E-CoV in Li et al. (2019), NL63-CoV in Walls et al. (2016), and HKU1-CoV in Kirchdoerfer et al. (2016). These are summarized in Supplementary Table S1. The relevant triple of chains of each monomer is given, as well as the conformation up/down of each of their heads expressed as u/d; if the corresponding head is only partly up, but more up than down, then this is expressed as u′ with a similar interpretation of d′. It may well be worthwhile to quantify the extent of up/down for future investigations. It is worth explaining that the MERS-CoV 5W9*-series of examples, that is 5W9H-5W9P, are bound to an engineered antibody G4 that dimerizes the spike, hence the two triples of spike chains in these cases.
Chosen for further initial analysis are representative structures, one for each hCoV spike: SARS-CoV-1 (5X58), SARS-CoV-2 (6VXX), MERS-CoV (6Q04), NL63-CoV (5SZS), OC43-CoV (6OHW), HKU1-CoV (5I08), and 229E-CoV (6U7H). These were chosen since they are all in the d-d-d configuration and are as wild as possible though stabilization in the prefusion conformation demands some form of experimental intervention.
There is the potentially interesting aspect that the up/down configuration of the heads is reflected in the backbone free energy. If so, then by appropriately targeting free energy sites to freeze the heads either up or down, one might incapacitate receptor binding in the down position, or render both receptor binding domain and fusion peptide susceptible to immune system or other attack in the up position. Even simply changing the relative frequencies of up/down could interfere with binding or facilitate attack and prevent serious infection. A different approach is taken here, but this explains the demand that all files for further initial comparison be in the d-d-d configuration to achieve free energy profiles that are as comparable as possible; only this configuration is available in the PDB for endemic hCoV diseases.
2.4. Bifurcated backbone hydrogen bonds
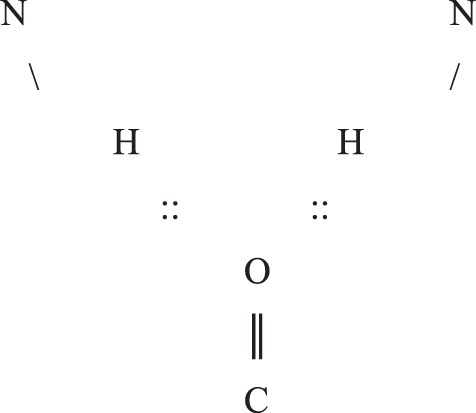
Hydrogen bonding occurs when an electronegative atom, such as O, approaches another electronegative atom, such as N, which is bound to an H, and the two in effect share the electron cloud of H. N is the donor and O the acceptor of such a hydrogen bond. These are short range interactions, ideally on the order of 3 Å from N to O, and in an aqueous environment, they lie near the bounds of stability owing to entropic effects. See Finkelstein and Ptitsyn (2016) for detailed discussion. The salient example here consists of the protein backbone atoms C = O and N-H from different peptide groups, which may participate in a hydrogen bond C = O::H-N called a backbone hydrogen bond. It can sometimes happen that a single O accepts two such backbone hydrogen bonds, called a bifurcated hydrogen bond, as depicted in the diagram and rarely can be trifurcated and participate in three. A single N-H likewise only rarely donates to more than one hydrogen bond. From a certain point of view, bifurcated hydrogen bonds of any type crudely reflect gross quantum effects.
3. Methods
There are several steps in the current analysis as follows:
4. Results
The purely data-intensive methods result in the sites of interest listed in Supplementary Table S3, namely, the five residues 131, 203, 392, 1029, and 1058 for SARS-CoV-2 listed across the first row. The hydrogen bond donor residues to 131 are 117/134, to 203 are 227/228, to 392 are 523/524, to 1029 are 1034/1035, and to 1058 are 730/731. Table 1 enumerates all such tuples of residues comprising the sites of interest in the spikes discovered here and aligned across all hCoVs.
Aligned Sites and Solvent-Accessible Surface Areas
Aligned groups of residues comprising sites of interest are given across all seven human coronaviruses. The first residue in each tuple is bifurcated or trifurcated. Below the residue numbers are given the one-letter residue codes of primary structure along with the solvent-accessible surface areas in square Angstrom taken from the Dictionary of Secondary Structure for Proteins introduced in Kabsch and Sander (1983). In boldface are given those sites that are exposed.
Before discussing Table 1, there is much that is parenthetically interesting in Supplementary Table S3 as is briefly examined next. For example, the 6AC*-series of low pH experiments for SARS-CoV-1 show that the aligned bifurcation at residue 1010–1011 (one or another was bifurcated over the population of SARS-CoV-1 files) is destroyed by low pH, whereas the others are not. Since the endocytic pathway is itself acidifying, this suggests that this particular bifurcated high free energy bond may be broken in postbinding fusion reconformation. Also, the disappearance of the bifurcation in the 5X5*-series for MERS-CoV suggests that the S2-cleavage site mutation destroys the bifurcated bond at residue 432. Because of the dimerization in the 5W9*-series for MERS-CoV in the presence of the engineered antibody, it is not clear whether the potential wealth of up/down data reliably reflects the situation in vivo as reflected in the odd shift of bifurcation of residue 174, for example. Note that, remarkably, residue 174 for MERS-CoV is actually typically trifurcated with maximum free energy for all three constituent hydrogen bonds, an extremely exotic residue indeed. It is likely that the different experimental techniques used to stabilize the spike in its prefusion conformation might subtly affect the free energy profiles, although it has been shown in Kirchdoerfer et al. (2016) that the so-called 2P two-proline substitution noted in Supplementary Table S1 is relatively inconsequential from other points of view.
More generally, it was already mentioned that in an another approach to antiviral targets, one could search for free energy patterns to explain the up/down configurations of the heads. Perusal of Supplementary Table S3 shows that such a signal, if it exists and depends only on the residues specified in the table, is not so simple and may involve the free energy at several sites across all three chains.*
The main conclusion of Supplementary Table S3 at this moment, though, is that 3D neighborhoods of SARS-CoV-2 residues 131, 203, 392, 1029, and 1058 provide potential sites of interest, as already discussed. There remains the issue of whether these residues are accessible; that is, do they lie exposed in the spike molecular surface?
Supplementary Figure S1 (overviews) and Figure 1 of the main text (close-ups) demonstrate that residues 131, 203, and 1058 are indeed exposed, whereas residues 1029 and 392 are not. Actually, it is more interesting, since it is presumably the bonds, hence pairs of residues, which may or may not be vulnerable, and in this case of bifurcated bonds, it is triples of residues that are liable. However, this does not rescue residue 1029, since its partner residues 1034 and 1035 are also not accessible, but there is a partial rescue of residue 392, since one of its partners, namely residue 523, is somewhat accessible, but only when the head is in the up conformation.

Close-ups of exposed residues for SARS-CoV-2:
Returning to the remark in Section 3 Step 5 and scanning Supplementary Table S3, one sees that it is only NL63-CoV residue 1097 and 229E-CoV residues 312 and 318 that fail to be bifurcated, and these align with residues in SARS-CoV-2 that are not accessible. One, therefore, could have eliminated this exception in Step 5, demanding all examples to be bifurcated, arriving at the same conclusions. Alternatively, one could have simply excluded in Step 5 any residues that are not accessible for SARS CoV-2. However, these two exceptional cases exhibit important phenomena in Supplementary Table S3, and, therefore, were included despite the seemingly arbitrary condition in Step 5, which could have been abandoned.
5. Discussion
The approach succeeds and identifies sites of interest for SARS-CoV-2 satisfying all the stated requirements. Interestingly, one porcine CoV spike glycoprotein (6U7K) given in Wrapp and McLellan (2019) was analyzed, and none of SARS-CoV-2 residues 131, 203, and 392 found analogues there, but residues 1029 and 1058 did; one murine CoV spike glycoprotein (6VSJ) given in Shang et al. (2020) was also analyzed, and again none of SARS-CoV-2 residues 131, 203, and 392 found analogues there (and the receptor binding site is on the lobe not the head), nor 1058, but residue 1029 did. This likely reflects differences between host species for CoV recognition and binding, but similarities for fusion.
The triples of aligned bifurcated residues in the chosen structures are given in Table 1 for all hCoVs along with the corresponding triples of primary structure and solvent-accessible surface area. Solvent-accessible surface area alone does not fully describe exposure for certain sites with high accessible surface area may still be interior to large cavernous recesses of the spike, whereas sites without it might lie in modest-sized canyons or troughs. Among the hCoVs, the three accessible sites on the SARS-CoV-2 spike seem especially exposed; see Supplementary Figure S2 for overviews of the other hCoV spike glycoproteins. Two of the exposed sites for SARS-CoV-2 lie on the lobes, site 1 being distal to the nearby head with site 2 more proximal to the virus envelope than site 1, and with site 5 still more proximal than the head/lobe region.
It is important to stress that all structures considered here have been modified in some manner to be stabilized in the prefusion conformation. The sites of interest, therefore, distinguish regions whose high free energy has not yet been released for conformational change. In particular, the sites on the lobes are not involved in receptor binding as one can readily verify from the cocrystal examples and are likely blocked from their further function by stabilization.
Other approaches to SARS-CoV-2 using the technology here are conceivable. Already mentioned is the possibility of finding free energy signatures for the up/down configurations of the heads, and interfering with or altering their frequencies (cf. Note added in proof). However, note that the permutation of chains between heads and lobes mentioned before suggests this signature, if such there be, could likely span all three chains. Furthermore, the backbone free energies for PDB files of viral structures other than the full spike might also be analyzed for sites of interest.
In any case, the difficulties of promoting sites of interest to vaccine targets (cf. Rueckert and Gusmán, 2012), to drug targets (cf. Mandal et al., 2009), or to effective tests for infection are manifold and substantial. Only two points in this regard will be mentioned: perhaps small molecules or nanobodies (Wesolowski 2009) might provide more auspicious obstructive vehicles than antibodies, since the immune systems of the many casualties so far from COVID, and the vast number of failed challenges of their own immune systems indicate that Mother Nature, with her wider vocabulary of sites and compounds, has not succeeded in finding antibodies, so how can we expect greater success? And the backbone coding advocated here is not only more resilient to viral mutational drift but also better shielded from adaptive recognition.
Only laboratory experimental work can be expected to confirm or refute the utility of what has been established here. It is, therefore, imperative to quickly proceed to laboratory verification! The details of this will be taken up elsewhere.
It should not escape the notice of the reader that sites for other viruses, and indeed for other types of diseases and biological processes, might be discovered in this same manner.
Footnotes
Acknowledgments
It is a pleasure to thank Minus van Baalen, Misha Gromov, Pablo Guardado-Calvo, Willi Jäger, Nadya Morozova, Michael Waterman, and especially Arndt Bennecke for valuable discussions, and François Bachelier and Greg McShane for vital computer assistance.
Author Disclosure Statement
Dr. Penner is a chair at IHES. No competing financial interests exist.
Funding Information
No funding was received for this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.