
Abstract
MicroRNAs are involved in many critical cellular activities through binding to their mRNA targets, for example, in cell proliferation, differentiation, death, growth control, and developmental timing. Prediction of microRNA targets can assist in efficient experimental investigations on the functional roles of these small noncoding RNAs. Their accurate prediction, however, remains a challenge due to the limited understanding of underlying processes in recognizing microRNA targets. In this article, we introduce an algorithm that aims at not only predicting microRNA targets accurately but also assisting in vivo experiments to understand the mechanisms of targeting. The algorithm learns a unique hypothesis for each possible mechanism of microRNA targeting. These hypotheses are utilized to build a superior target predictor and for biologically meaningful partitioning of the data set of microRNA–target duplexes. Experimentally verified features for recognizing targets that incorporated in the algorithm enable the establishment of hypotheses that can be correlated with target recognition mechanisms. Our results and analysis show that our algorithm outperforms state-of-the-art data-driven approaches such as deep learning models and machine learning algorithms and rule-based methods for instance miRanda and RNAhybrid. In addition, feature selection on the partitions, provided by our algorithm, confirms that the partitioning mechanism is closely related to biological mechanisms of microRNA targeting. The resulting data partitions can potentially be used for in vivo experiments to aid in the discovery of the targeting mechanisms.
1. Introduction
MicroRNAs are short RNA sequences of ∼22
Despite the importance of microRNAs, the detailed mechanism of microRNA target binding is poorly known. Laboratory experiments for finding targets are very slow and costly; therefore, there is a huge demand for computational approaches. In the last decade, dozens of algorithms, with a variety of approaches and techniques, have been developed. These methods are either specific for a few species or general for any kind. Methods for vertebrates include TargetScan and TargetScanS (Lewis et al., 2003, 2005), miRanda (Enright et al., 2004; John et al., 2004), DIANA-microT (Kiriakidou et al., 2004), and for flies RNAhybrid (Rehmsmeier et al., 2004). Some general tools are miTarget (Kim et al., 2006) and MicroInspector (Rusinov et al., 2005).
The early computational approaches for target recognition were rule based, that is, they had a set of discriminative rules derived from experimental and biological knowledge, such as minimum free energy (MFE), duplex binding pattern, or target accessibility. Some popular rule-based tools are RNAhybrid, TargetScan, miRanda, and MirBooking (Weill et al., 2015). MirBooking is one of the recent rule-based methods that simulate the microRNA and mRNA hybridization competition and cellular conditions to improve the accuracy of target prediction. In the last several years, with the increase of relevant data sets, data-driven methods have been attempted. These methods use sophisticated machine learning (ML) and statistical models to learn more discriminative features for target identification (Yue et al., 2009). Some popular data-driven tools are TargetSpy (Sturm et al., 2010), miRanda-mirSVR (Betel et al., 2010), and Avishkar (Ghoshal et al., 2015). However, such methods have yet to resolve the issue of high false-positive rate. The innovation of more advanced sequencing techniques, and therefore more precise data sets, along with recent advances in ML methods, could lead to the development of more accurate algorithms.
The microRNA targeting process has not been well understood; biologists are especially interested in approaches that may provide insights about the mechanisms of target recognition. Recent experimental studies of microRNA targeting reveal that there are multiple and different mechanisms for this process, whereas the earlier belief was merely based on seed match of microRNA and target site sequences (Cloonan, 2015). Currently, it is still not clear how many different and exclusive mechanisms guide microRNA targeting; therefore, computational models, which not only work well but also give insight into the biological mechanisms, are very desirable. Some ML techniques such as Bagging and Boosting or Random Forest aim to learn multiple hypotheses from the input data, but they do not provide any clue to check if these hypotheses are biologically meaningful or not. Biologically meaningful features here mean those characteristics that have been experimentally confirmed to be part of a microRNA targeting mechanism, such as appearance of adenine at the far 3′ end of a target site.
In this article, we introduce a multi-hypotheses learning (MHL) algorithm (Mohebbi et al., 2019) that aims at not only improving the performance of classifiers on microRNA targeting data but also meaningfully partitioning the data set per these learned hypotheses. MHL uses these hypotheses to predict microRNA targets with a superior performance. Moreover, the biologically meaningful partitions created by MHL could be used for further understanding the targeting mechanisms or to discover new target determinants. To verify our approach, we evaluated our method on human and mouse data. The results show that the partitioning is indeed biologically meaningful. Moreover, significant performance improvements on target prediction confirm learning multiple hypotheses can help outperform top ML algorithms such as RandomForest and deep learning (DL) models. Feature analysis of the partitions produced by MHL reveals interactions in microRNA and target duplex that are verified by the biology literature. This supports our conjecture that MHL could aid to mine meaningful features, which could be used as part of in vivo experiments.
2. Data Sets
The success of data-driven methods critically relies on the quality of the data. To build the most accurate models and the most realistic evaluations, we extracted our data from mirTarBase (Hsu et al., 2014), one of the most up-to-date data sets and the most referenced resource for microRNA target prediction research. In particular, mirTarBase contains more than 360,000 experimentally validated microRNA–target duplexes from 18 different species. This data set is publicly available and is subject to IRB Waiver. We are mostly interested in testing our ML method with both human and mouse records.
From mirTarBase, human and mouse microRNA–target duplexes were extracted whose secondary structures have been provided in research articles. Such duplexes were selected as positive samples for our method. However, negative samples are not directly available. Theoretically, any stretch of an appropriate length other than the real target in the 3′-UTR of a targeted mRNA gene can be considered a negative target of the corresponding microRNA. We randomly selected 10 locations in the 3′-UTR of a targeted mRNA gene to pick up the negative samples for each positive sample with a ratio of 10:1. Each sample is a pair of microRNA sequence of length 22 and a site sequence of length 25, which is real target site for positive samples or a negative site that is not a target for the microRNA.
2.1. Test set and training set
We have one training set and two test sets. The human data set is split 80% to 20% into the training set and human test set. All mouse data compose our second test set. In the human data extracted from mirTarBase, there are 322 unique microRNAs, 3651 target site sequences, and 3722 pairs of microRNA and target sites. On average, each microRNA has >10 targets sites. If we randomly select test set samples from the whole database, the odds of having many microRNAs in both test and training sets is high. To avoid such overlaps and to have the most reliable test set, we indexed pairs of microRNA and target sites by microRNA sequence. In addition, to make a test set with a similar distribution to that of the whole data set, we sort samples by microRNA sequences, put four consecutive (based on the sorting order) microRNA sequences and all their target and nontarget sites in the training set, then one microRNA sequence and all its targets and nontargets into the human test set, and so on. In this way and in terms of microRNA sequence, not only do the human test set and training set have no overlaps but also the test set has very similar distribution to that of the whole database. Both test sets and training sets have ratio of 1:10 for positive versus negative. The human test set consists of 6127 samples (557 positives vs. 5570 negatives), and the total size of the mouse test set is 517.
3. Materials and Methods
In this section, we introduce a feature selection approach that is biologically meaningful and is more efficient for microRNA targeting than common data mining feature selection methods. Data mining algorithms may not be applied directly to this problem because each sample is composed of sequences of microRNA and target, and the microRNA sequence is identical among its positive(s) and its negative samples. To verify this thought, we ran Weka (Witten et al., 2016), a data mining package, to extract the most significant features from samples of a microRNA sequence concatenated to a target sequence. All microRNA sequence nucleotides were excluded from selected features set and that is because the microRNA sequence is the common part of both positive and negative samples; therefore, feature selection algorithms consider the common part as a none determinant factor for classifying the samples. To cope with this problem, features must be defined based on correlations of microRNA and its target nucleotides rather than merely on sequences of nucleotides. In addition, and to incorporate biological knowledge of microRNA targeting, we extracted features from the secondary structure of a duplex associated with every sample.
We researched on the most recent discoveries on the mechanisms of microRNA targeting and customized RNAfold (Lorenz et al., 2011), a widely used secondary structure prediction tool for RNA sequences, to predict a specific structure for a pair of microRNA and target sequences that mimic the biological mechanisms of microRNA targeting. RNAfold is a general tool for secondary structure prediction, and it could predict structures in which microRNA or target site sequences bind to themselves or to each others in any ways. On the other side and biologically, sequences of microRNA and target sites should not make base pairs with themselves but with the other sequence. To guide RNAfold prediction for the purpose of microRNA targeting and include information about in vivo process of microRNA target binding, we tuned RNAfold to predict the structure of duplex based on rules we collected from biological literatures explaining the actual mechanisms of microRNA targeting.
The seed part of a microRNA consists of the nucleotides number 2 to 8 from the 5′ end of the microRNA (Lewis et al., 2003). It is believed that the process of nucleotide binding between the microRNA and its mRNA target starts from this region (Schirle et al., 2014). When the binding in the seed region is continuous for 6 to 8 bps, it is called a canonical seed; otherwise, it is called noncanonical (Loeb et al., 2012). Although the seed binding is considered the most important identifier for microRNA targets in mammals (Peterson et al., 2014), a recent study shows that it is not the only mechanism for microRNA targeting (Cloonan, 2015). To have a more comprehensive model, we considered correlations that occur not just in the seed region but also in all other regions across a microRNA and its target.
3.1. MicroRNA duplex secondary structure prediction
RNAfold predicts the most stable secondary structure of a single RNA sequence. To use it for predicting microRNA and target site duplexes, we concatenated microRNA and target site sequences with a subsequence of length 4 “X”s in between, as shown in Figure 1. This sequence of length 4 is the shortest sequence that we could add and still get the same MFE for the structure as the MFE we get from RNAcofold (Lorenz et al., 2011) when it predicts the duplex between microRNA and target site.
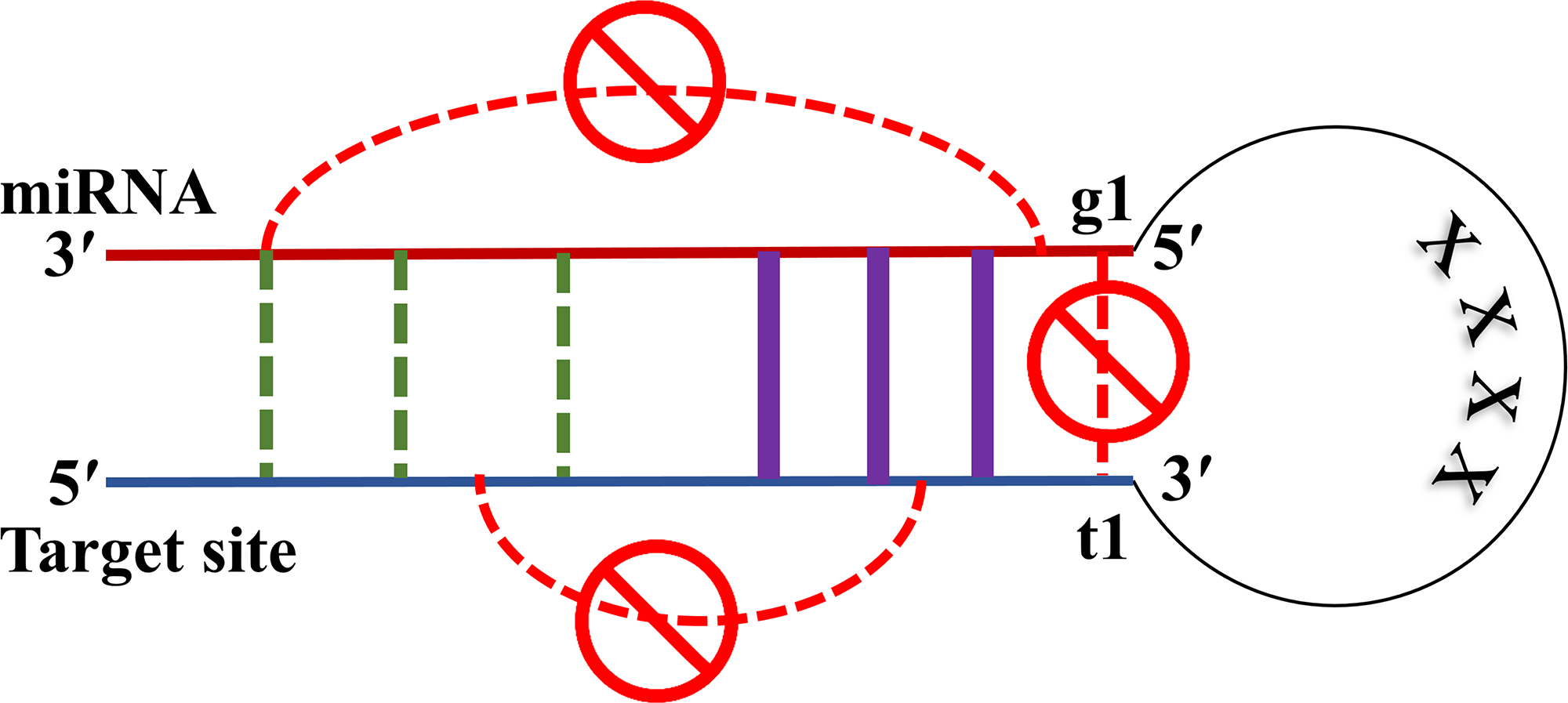
RNAfold customization for a microRNA (miRNA) –target duplex; sequences of microRNA and target site are concatenated with a subsequence of length 4 “X”s in between. Base pairing among nucleotides of one sequence is prohibited, either for microRNA or for the target site. A structural study on the mechanism of microRNA targeting (Schirle et al., 2014) reveals that the nucleotide in t1 goes into a pocket inside the Argonaute protein structure and does not pair with the corresponding nucleotide on microRNA. We added a constraint to prevent t1 from such a base pairing with the microRNA. Base pairs (purple color) are enforced through the customization if the corresponding nucleotides are complimentary matching. Black dashed lines show possible locations of valid base pairs, and we let RNAfold to predict them.
RNAfold can have a constraints file as an input parameter to enforce the structure prediction process to occur based on a user's domain knowledge. Here, we set these constraints for the microRNA targeting mechanism, to include rules for base pairs that are biologically expected to happen in seed, and rules prohibiting microRNA nucleotides from binding to microRNA itself. Similarly, there are rules avoiding target site sequence to bind over itself. We applied all these rules for duplexes with canonical seeds, while releasing seed base pairing constraints for noncanonical seeds.
Biological experiments and in vivo methods reveal several mechanisms for microRNA targeting (Cloonan, 2015). The earliest discovered and the most dominant method of targeting was based on seed matching (Lewis et al., 2005; Friedman et al., 2009). In this mechanism, microRNA carried by the Argonaute protein makes initial base pairs in the seed area. These bindings open the groove of Argonaut molecule to accommodate the target site (Schirle et al., 2014). To customize RNAfold for predicting duplexes in a similar manner, we aligned the seed part of microRNA with nucleotides 2 to 8 from 3′ side of target and pair these bases that can match to each other mutually, that is, adenine (A) to uracil (U), cytosine (C) to guanine (G), guanine to uracil, and vice versa.
3.2. Numericalization of a microRNA duplex structure
RNAfold predicts a secondary structure as a list of base pairs between nucleotides in a microRNA and its target site. To apply ML-based algorithms on the structure, we needed to convert it to a vector of numbers. These base pairing features are nominal and to convert them into numerical values while maintaining their independence, we encoded them with the one-hot-encoding approach (Sujit Pal, 2017). Biologically, there is no significance to the ordering among six different base pairs; to keep this independence, we encoded each matching base pair with one bit, totaling six bits, and one extra bit for mismatches or no base pairs. One and only one of these seven bits is hot or one at any time. In a microRNA duplex structure, there might be bulges on either microRNA or target sequence. To numericalize this information and add it to the vector, we assigned two integer values indicating size of bulges on the microRNA and on the target site, adjacent to each nucleotide. These values are zero if there is no bulge in the structure next to the current nucleotide. In total, there are nine features per each microRNA nucleotide.
Experimental studies on human microRNA targets showed that adenine is a very frequent base at the far 3′ end of a target site, that is, at t1 (Schirle et al., 2014; Lewis et al., 2005). To add this biological preference to features set, we added four bits corresponding to A, C, G, and U at t1. A study on the structural basis of microRNA targeting (Schirle et al., 2014) revealed that the nucleotide in t1 goes into a pocket inside the Argonaute protein structure and does not pair to the corresponding nucleotide on microRNA, that is, g1, which is the first nucleotide on 5′ end of the microRNA. Therefore, to reduce the size of features set, we excluded g1 from being encoded. A factor indicating stability of a structural binding is MFE, we included it as the last feature. We fixed the length of microRNAs to 22 nucleotides, but g1 is not considered; therefore, the total number of features for each sample is 194 or (1 + 4 + 21*9). If the length of microRNA is larger than 22, the sequence is trimmed to 22 from 3′ side of microRNA. This procedure is illustrated inside dashed area in Figure 2. Our MHL algorithm, to be introduced in the next section, treats each sample as a vector of these 194 features and learns several hypotheses each corresponding to a different microRNA targeting mechanism. Figure 2 shows all components of our bundle algorithm including the feature selection part and the MHL algorithm.

Our bundle algorithm; it gets two sequences of microRNA and target, and concatenates them with subsequence “XXXX.” The resulting sequence is passed to RNAfold that we customized as explained in Figure 1. The customized RNAfold predicts the secondary structure of the microRNA and the target duplex. The structure is encoded as a vector of features and passed down to MHL (shown in Fig. 3). MHL, multi-hypotheses learning.
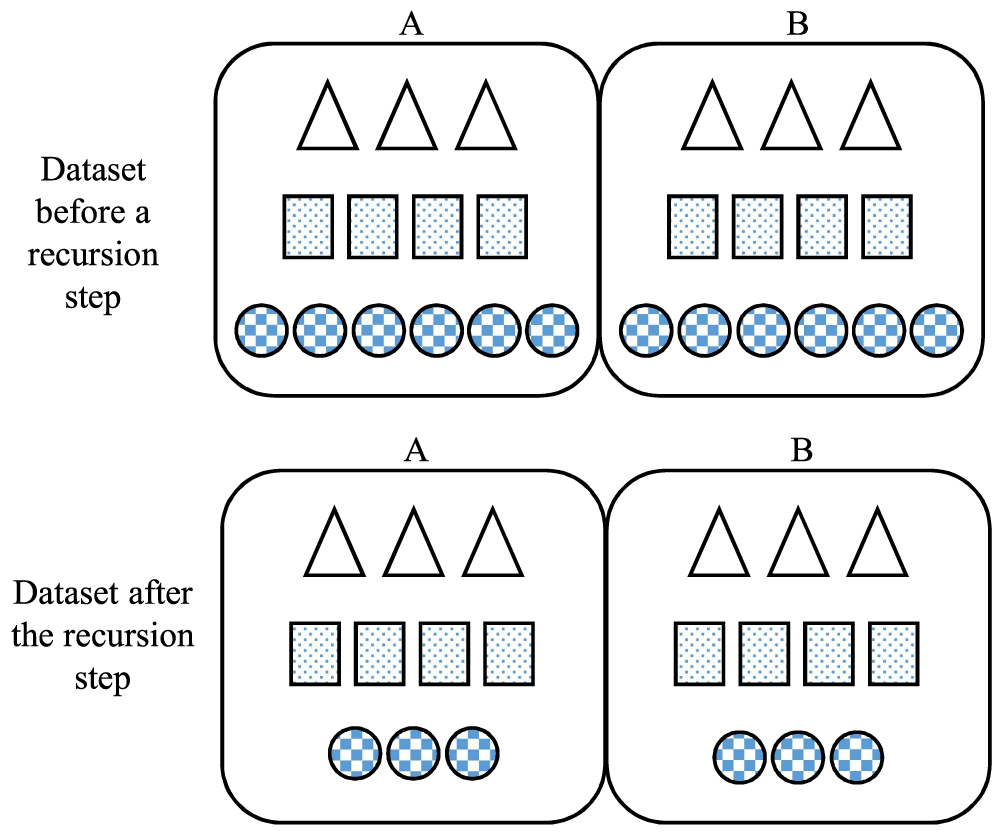
The illustration of the MHL recursion algorithm; data set D is split to subsets A and B, and then, a classifier ci is trained on A. It captures the dominant pattern, here circles. The trained model can detect the similar pattern in B, that is circles; these form the
3.3. An MHL algorithm
The idea of the algorithm is to divide the data set into two disjoint subsets
The algorithm consists of two main parts: Trainer and Tester. Trainer gets the training set T0, a classifier set C, and a desired sensitivity and specificity:
3.4. Trainer
Trainer consists of three functions:
Depending on how high the thresholds
There are two thresholds associated with each trained model;
3.5. Tester
The Tester procedure loads all model files, mi's, from the training step into the memory, and when a new and unlabeled sample is given for evaluation, all models examine the sample. If a model evaluates the sample with a value between
4. Results and Discussion
In the training set, there might be several patterns of microRNA targeting, here we denote them symbolically by circles, squares, triangles, and so on, as an example shown in Figure 3. Initially, circles are the dominant pattern. The MHL algorithm divides it into subsets A and B. Classifier c learns circles pattern when it runs over subset A and creates a model for circles, that is, mc. Evaluating B with mc divides B into two partitions; samples decidable by mc, that is, the
4.1. Comparison with a DL model of microRNA–target duplexes
DL, a.k.a deep neural networks (NN), is a multilayer artificial NN that can learn complex functions and correlations among input variables. It has been used for microRNA target prediction in several recent methods such as miRAW (Pla et al., 2018) and DeepTarget (Lee et al., 2016). To have a fair comparison based on the same training set and test sets, we built our deep neural network with three layers. In the first layer, there are 4 × 22 × 25 = 188 input nodes, where 22 = length of microRNA sequences and 25 = length of target sequences; we used the one-hot-encoding approach (Sujit Pal, 2017) to convert nominal inputs to numerical values to provide input to the network. The second layer, that is, the hidden layer, has the same number of notes as the first layer. The third layer has one node as the output of the network. We used Keras (Chollet et al., 2015) and TensorFlow to implement our DL model and used grid search for the optimum parameter selection. The training set was split into 70% and 30% for training and validation, respectively. The best performance on the validation set was achieved by parameters “
4.2. Test of our MHL algorithm
To test the effectiveness of our algorithm, we measure the area under the precision–recall (PR) curve (AUPRC) and the area under the receiver operating characteristic (ROC) curve (AUC). The AUC has been widely used to measure the performance of a binary classifier, and the AUPRC could provide better metrics to compare classifiers on an imbalanced test set (Boyd et al., 2013). A random classifier has AUC = 0.5 and
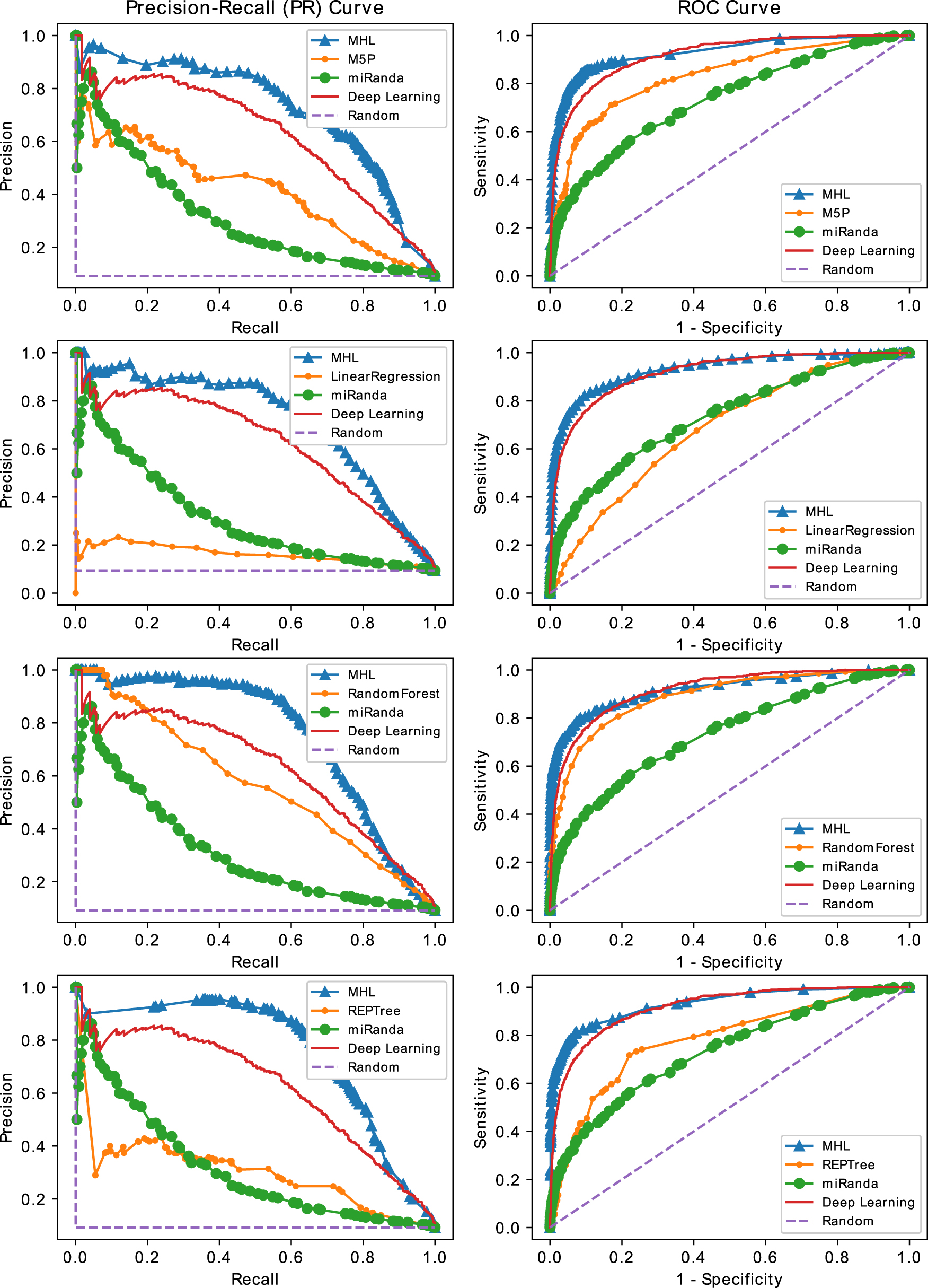
PR and ROC curves of MHL versus ML models, miRanda, a deep learning model and random classifier on HSA (Human) test set. Each row of plots depicts the performance comparisons of MHL versus an ML model in terms of PR and ROC curves. The ML algorithms from the top are M5P, LinearRegression, RandomForest, and REPTree, respectively. The highest improvement due to MHL could be seen in the second row plots when MHL uses LinearRegression as a base classifier. ML, machine learning; PR, precision–recall; ROC, receiver operating characteristic.
The Area Under the Receiver Operating Characteristic Curve and the Area Under the Precision–Recall Curve of Different Machine Learning Models Versus our Multi-Hypotheses Learning Algorithm on HSA (Human) Test Set,
AUPRC, area under the precision–recall curve; AUC, area under the receiver operating characteristic curve; MHL, multi-hypotheses learning; ML, machine learning; NN, neural network.
The Area Under the Receiver Operating Characteristic Curve and the Area Under the Precision–Recall Curve of Machine Learning Models Versus Our Multi-Hypotheses Learning Algorithm When They Were Trained on Human and Tested on MMU (Mouse) Test Set,
These tables show that our algorithm is effective, and MHL always achieved a superior performance over ML models. We compare the performances in terms of both AUPRC and AUC. Performance of a binary classifier on balanced data sets is similar in terms of AUPRC and AUC, while in imbalanced test sets, AUPRC is a better determinant of performance. Our test sets have a ratio of 1:10 for positives versus negative samples; therefore, AUPRC values should represent better evaluations for the performance of models.
In Table 1 where models are tested on Human samples, the highest overall performance is achieved by MHL, when it uses REPTree as a base classifier. The model trained by REPTree itself on our training set has a performance of AUC = 0.90 and AUPRC = 0.60. MHL, by using REPTree as base classifier and learning multi-hypotheses, increased the performance to AUC = 0.93 and AUPRC = 0.76. By comparing these measures to those of a random classifier, that is, AUC = 0.50 and AUPRC = 0.09, we can observe that MHL is successful in distinguishing samples that represent a microRNA target from the samples that contain nontargets. The last two columns show improvements achieved by MHL over the base classifiers; the highest improvement in terms of both AUC and AUPRC is when LinearRegression is used as the base classifier.
On our human test set, LinearRegression alone has a mediocre performance with AUC = 0.68 and AUPRC = 0.16. MHL utilized the same classifier to achieve a very high performance with AUC = 0.93 and AUPRC = 0.72. The improvement is 0.25 in AUC and 0.56 in AUPRC. The best performing classifier, trained without MHL, is DL NN with a performance of AUC = 0.91 and AUPRC = 0.71. The second is RandomForest with AUC = 0.89 and AUPRC = 0.57. Classifiers, performing poorly on this test set such as RandomTree and DecisionStump with AUPRCs 0.26 and 0.29, can also be used in MHL to deliver a performance of 0.54 and 0.58. The performance of classifiers alone has an average AUC of 0.71 with a standard deviation 0.13, and an average AUPRC of 0.35 with a standard deviation 0.13. For MHL, the average AUC is 0.87 with a standard deviation of 0.08, and the average AUPRC is 0.69 with a standard deviation of 0.09. MHL increased AUC by 22.53% and AUPRC by 97.14% on average. MHL almost doubled the performance of classifiers in terms of AUPRC. Improvement in AUC is not as high as AUPRC because AUC values for MHL are near perfect with 0.93 for four of the seven classifiers.
We also compare the models trained on our data sets versus two popular algorithms for microRNA target prediction miRanda (Betel et al., 2008) and RNAhybrid (Rehmsmeier et al., 2004). miRanda has an average performance, but RNAhybrid is unable to distinguish targets from nontargets accurately, and that is because miRanda and RNAhybrid are rule-based methods while other models are data-driven. Rule-based methods use microRNA–target duplex stability and its MFE to detect targets. MicroRNA duplexes with noncanonical seeds tend to have higher MFE than duplexes with canonical seeds, due to mismatches in the seed area (Loeb et al., 2012). RNAhybrid uses MFE of the duplex for target recognition, and it does not work well if test set has targets containing noncanonical seeds or has nontargets with relatively low MFE. On the other side, data-driven models could learn more complex correlations between nucleotides in microRNA and target, beyond the MFE and secondary structure of the duplex. This gives an advantage to data-driven models over these rule-based methods.
Table 2 presents the results of training models on Human data and testing on mouse samples. Human and mouse branched from a common ancestor about 80 million years ago. They have similar genomes and virtually the same set of genes (Genome.gov, 2017). Therefore, it is of interest to train a model by human genomic data and test it on mouse data sets. We ran the same model used for testing human data on mouse data set. Our algorithm improves over all ML classifiers, and the maximum improvement again is for Linear Regression with an increase of 0.45 in AUC and 0.82 in AUPRC. The average AUPRC of our algorithm is 0.85 with a standard deviation of 0.16, and for the other ML models that is 0.44 with a standard deviation of 0.25. The MHL algorithm average performance surpasses other ML methods average by 23.43% in AUC and 95.42% in AUPRC.
MHL has similar performance on both mouse and human test sets and that suggests that microRNA–target duplexes and targeting mechanism features are evolutionary conserved across both species. Some microRNAs have conserved sequences among humans and mouse; therefore, there might be a small set of samples with similar sequences in both the (human) training set and the mouse test set. This could be the reason for larger performance improvement on the mouse versus the human test set in Tables 1 and 2, respectively. We reduced the chance of sequence similarity in the human test set by sorting microRNAs and dividing them between training and test sets alternatively with the given ratio of 10:1, and also we subtracted the test set from the training set to avoid any chance of overlaps between the two sets.
Figure 4 shows the PR and ROC curves of our tests. In each row of the figure, there is a pair of plots that show the performance of MHL versus an ML model, miRanda, a DL model and random classifier, when they are evaluated on the human test set. The first row from the top shows performance of M5P ML model versus MHL when it uses M5P as a base classifier. The difference between MHL and M5P curves clearly shows the effectiveness of our algorithm; the M5P curve reflects the performance of the single hypothesis learned by the model, and MHL curve represents the predictive power of several hypotheses learned by MHL. The closest competitor to MHL is the DL model that we trained on our training set. While the performance of the DL model is due to the power of tool and our grid search parameter selection strategy, the superior performance of MHL comes from simulating the structural mechanism of microRNA targeting and also learning hypotheses that model different mechanisms of targeting. Rows 2, 3, and 4 show similar comparisons for MHL versus LinearRegression, RandomForest and REPTree, respectively. The difference in MHL curves, in each row, comes from the different base classifiers that were used within MHL to learn multiple hypotheses. While the performance of ML models varies significantly among themselves, MHL consistently surpassed them and even the powerful DL model by a high margin.
While some data-driven methods, such as RandomForest and DL models have comparable performance with AUC of 0.89 and 0.91, respectively, or GraB-miTarget (Mohebbi et al., 2018), a hybrid model of a graph and a support vector machine, has an AUC performance as high as 0.92, the distinctive advantage of MHL is to provide a clue into the data by partitioning the data into subsets that contain biologically related microRNA duplexes.
4.3. Feature selection analysis of subsets provided by MHL
To check if the samples within subsets provided by MHL have biologically meaningful correlation, we select significant features within each subset and also within the entire training set by using correlation-based feature selection (CFS) algorithm (Hall, 1999), and then, we compare features mined in each subset versus features extracted from the entire training set. From the Weka package, the CFS algorithm was run; the features extracted from the training set and the subsets created by MHL are shown in columns 1 to 6 of Table 3. In our notation for features' name, i is the nucleotide index starting from 1 on 5′ side of a microRNA sequence, and the names are in the format of
Feature Selected by Correlation-Based Feature Selection, from Complete Training Set Versus from Five Subsets Partitioned by the Multi-Hypotheses Learning Algorithm
The comparison of first row with rows 2 to 6 shows that MHL can help to extract biological details from subsets while they could not be captured by the CFS method on the complete training set. In each feature, the number represents the nucleotide index starting from 1 on 5′ side of microRNA. For example,
CFS, correlation-based feature selection; MFE, minimum free energy.
In Table 3, column 1, there are several biological details that are missing. The appearance of adenine in the first position of the target, that is,
CFS has not been able to extract G:U base pairs from the entire training set, and they are not present in column 1, but with MHL, microRNA duplexes with G:U base pairs have been detected and grouped into subset 4. Column 1 shows that CFS on the entire training data set has been able to detect bulges only in positions 4 and 6, but with MHL, we can detect these bulges in indexes 2, 3, 4, 5, 6, and 7. MicroRNAs with bulges in indexes 2 and 3 are clustered by MHL in subset 2. Subset 4 contains microRNAs with no bulge in the seed area. Subset 1 has microRNAs with bulges in the rear half of the seed, positions 4, 5, and 7. GC is a strong base pair, and a biological mechanism of targeting proposed by Schirle et al. (2014) claims base pairing on positions 2, 3, and 4 make the groove inside Argonaut protein to open and to accommodate the target. MHL has been able to cluster samples related to this mechanism into subsets 1, 2, and 5, while column 1 does not include any GC base pair feature in microRNA duplexes.
The feature
Continuous pairing to the 3′ side of microRNA can compensate for single nucleotide bulges or mismatches in the seed region (Bartel, 2009). The first column does not show a significant presence of such pairs, but columns 1 to 4 have continuous base pairs and also have more individual pairs at the 3′ side of microRNA; subsets 1 to 4 contain samples with adjacent pairs at positions 19 to 22. Subset 2, in addition, contains two more continuous base pairs at indexes 16 and 17.
These biologically interpretable details seen in subsets 1 to 5 but missed in column 1 shows that the MHL algorithm can provide subsets of the data that have biologically correlated samples. Samples in these subsets can be further studied or experimented to determine new targeting mechanisms. Based on the current understanding on mechanisms of microRNA targeting, some subsets or features may not have a verified biological interpretation, but they can be used in in vivo experiments to discover new targeting mechanisms.
4.4. Data set clustering of MHL versus standard clustering algorithms
MHL clusters a data set based on a set of optimum hypotheses that it learns through the process of training and that is what makes MHL distinct from standard clustering algorithms. To contrast the clustering merit of MHL versus other standard clustering algorithms, we ran clustering algorithms from the Weka package such as Canopy, Cobweb, Expected Maximization, FarthestFirst, FilteredClusterer, and Xmeans (Witten et al., 2016) on our data set. Table 4 contains these results; column 1 lists algorithms and column 2 shows the number of clusters created by each algorithm. The clustering algorithms from Weka either create too many clusters or splits the data set into two large subsets.
This Table Compares Multi-Hypotheses Learning Clustering Performance Versus Other Standard Algorithms
We ran these methods from the Weka package, listed in the first column, on our data set. The second column shows the number of clusters created by each algorithm. These algorithms either created too many clusters or split the data set into two large subsets. To evaluate the relevance of samples to each other in a cluster, we then ran the CFS algorithm. If samples in a cluster are relevant, the CFS algorithm gives a high Merit score for the selected features in the cluster. The Merit score is between 0 and 1. For each algorithm, we computed the weighted average of Merit scores for the clusters created by the algorithm, and it is shown in column 3. By comparing columns 2 and 3 for other algorithms versus MHL, one can observe that MHL could give better clusters in terms of the number of subsets and the average Merit score for the subsets.
EM, Expected Maximization.
To see how good is a clustering, we use the Merit score computed by the CFS algorithm for the input data set and for the features it selects. If samples in a cluster are relevant, the CFS algorithm gives a higher Merit score than if the samples are not related. The Merit score is between 0 and 1; a high Merit score means a low correlation between the selected features and a high correlation with the sample label.
For each cluster provided by a clustering algorithm, we ran CFS and used the Merit score as an estimate for the relevance of samples in the cluster. Table 4, column 3, shows the weighted average of Merit scores for each algorithm. We computed the weighted average because the number of clusters created by different algorithms varies. Weight for each cluster score is the proportion of the cluster size to the total number of samples in the training data set. The summation of the weights for each clustering algorithm is one.
Table 4 compares the clustering performance of MHL versus standard clustering algorithms from the Weka package. The results in this table show that MHL clusters better in terms of the number of clusters it creates and the weighted average of Merit scores for the clusters. MHL creates five subsets with an average Merit score of 0.548. The only algorithms with better Merit scores are FarthestFirst with the score 0.56 and Canopy's average score is 0.629. FarthestFirst has a small advantage for the Merit score, but it created two large clusters. Canopy has the highest score, while it created 100 small clusters. MHL is the optimum balance for the number of clusters and the average Merit score.
MHL creates clusters by utilizing the learned hypotheses. For each hypothesis, MHL chooses those samples that the hypothesis could either accept or reject with confidence, a vote close to one or zero, respectively. Other algorithms, however, partition the data set and create clusters based on similarity in attributes' values. Attributes could have different correlations to the label, and this may mislead a clustering algorithm away from partitioning by the most predictive attributes. MHL exhaustively searches for the hypotheses with the highest predictive performance and eventually those optimum ones cluster the data set; thus, MHL created clusters that are biologically meaningful and lead to better predictive performance.
In the last two decades, dozens of algorithms for microRNA target prediction has been published. These methods vary based on the information they use, their accessibility, being rule-based or data-driven, and their fundamentals. For a fair comparison of our method versus state-of-the-art methods, we studied several of the renown tools, such as TargetScan (Agarwal et al., 2015), TarPmiR (Ding et al., 2016), miRBase (Griffiths-Jones et al., 2006), DIANA-microT (Maragkakis et al., 2009), miRanda (Betel et al., 2008), miTarget (Kim et al., 2006), RNAhybrid (Rehmsmeier et al., 2004), Avishkar (Ghoshal et al., 2015), TargetSpy (Sturm et al., 2010), miRWalk (Dweep et al., 2011), and miRanda-mirSVR (Betel et al., 2010).
Some of these algorithms use a variety of other information, in addition to microRNA duplex sequences, for target prediction. For example, TargetScan (Agarwal et al., 2015), miRanda-mirSVR (Betel et al., 2010), and DIANA-microT (Maragkakis et al., 2009) use sequence conservation across species, conserved or nonconserved microRNA family, and miRBase annotation. Our MHL target predictions rely only on sequences of a microRNA duplex. Comparing MHL with methods such as TargetScan would be technically not feasible because our collected data do not have the information used by TargetScan. The other challenge was that the source code or executable files for some methods are not available, and they are only accessible through their online websites, for instance, miRDB (Wang, 2008), miTarget (Kim et al., 2006), and miRWalk (Dweep et al., 2011).
Our test set composed of 6646 samples (HSA 6129 samples and MMU 517), and we could not submit the test set as thousands of online queries manually. The functionality of some of these methods is different from our method, for example, TarPmiR (Ding et al., 2016) finds targets across a given mRNA for a microRNA sequence, whereas our data samples are pairs of short microRNA and target site sequences, and MHL is about finding out if a pair bind to each other. From available software and methods for microRNA target prediction, we could only use those with downloadable source code or executable code. Moreover, for a fair comparison, such methods would also need to predict a microRNA target merely based on sequences of microRNA and target site. Software tools satisfying all these requirements were miRanda (Betel et al., 2008) and RNAhybrid (Rehmsmeier et al., 2004). These rule-based methods work based on fundamental principles of microRNA targeting mechanisms such as a lower free energy binding and a stable secondary structure duplex. These metrics have had reliable performance; therefore, miRanda and RNAhybrid are still widely used either solely or as core components of other algorithms such as miRanda-mirSVR (Betel et al., 2010) and miRanda-MiRBase (Maziere and Enright, 2007).
5. Conclusion
MicroRNAs are small RNA sequences that regulate genes and have important and diverse functions. Biologists are very interested to discover the functionality of microRNAs, and their functions may be correlated with the way they detect their targets. As a result, microRNA studies and specifically microRNA target prediction received a lot of attention, and many computational algorithms have been developed in the last two decades. In this work, we presented a multi-hypotheses learner (MHL) algorithm that aims for two purposes: first, to predict microRNA targets with high accuracy, and second, to help with discovering the mechanisms of microRNA targeting, by providing partitions of samples that biologically correlate with each other within a partition. These partitions can potentially be used for in vivo experiments to discover new mechanisms of microRNAs target recognition.
Our MHL algorithm has significantly improved the performance of some ML methods by correct partitioning of the data set; for example, LinearRegression, MultilayerPerceptron, and REPTree have average performances when they are trained alone on the data set, but MHL boosts their performance to have very high performances and outperform powerful algorithms such as DL models and RandomForest. Feature selection analysis on the partitions created by MHL reveals that the MHL partitioning mechanism is indeed biologically meaningful and partitions have verified features that could not be mined without using the MHL algorithm.
Footnotes
Acknowledgments
The authors would like to thank Dr. Cory Momany and Dr. Khaled Rasheed for their feedbacks and comments on this work.
Author Disclosure Statement
The authors declare they have no competing financial interests.
Funding Information
This work was supported in part by the NIH grant (Award No: R01GM117596), as a part of Joint DMS/NIGMS Initiative to Support Research at the Interface of the Biological and Mathematical Sciences, and NSF IIS grant (Award No: 0916250).