
Abstract
Phylogenomics—the estimation of species trees from multilocus data sets—is a common step in many biological studies. However, this estimation is challenged by the fact that genes can evolve under processes, including incomplete lineage sorting (ILS) and gene duplication and loss (GDL), that make their trees different from the species tree. In this article, we address the challenge of estimating the species tree under GDL. We show that species trees are identifiable under a standard stochastic model for GDL, and that the polynomial-time algorithm ASTRAL-multi, a recent development in the ASTRAL suite of methods, is statistically consistent under this GDL model. We also provide a simulation study evaluating ASTRAL-multi for species tree estimation under GDL.
1. Introduction
Phylogeny estimation is a statistically and computationally complex estimation problem, due to heterogeneity across the genome resulting from processes such as incomplete lineage sorting (ILS), gene duplication and loss (GDL), rearrangements, gene flow, horizontal gene transfer (HGT), and introgression (Maddison, 1997).
Much is known about the problem of estimating species trees in the presence of ILS, as modeled by the multispecies coalescent (MSC) (Kingman, 1982; Takahata, 1989). For example, because the most probable unrooted tree for every four species is the species tree on those species (Allman et al., 2011), the unrooted species tree topology is identifiable under the MSC from its gene tree distribution, and quartet-based species tree estimation methods that operate by combining gene trees [such as BUCKy-pop (Larget et al., 2010) and ASTRAL (Mirarab et al., 2014; Mirarab and Warnow, 2015; Zhang et al., 2018)] are statistically consistent estimators of the unrooted species tree topology (i.e., as the number of sampled genes increases, almost surely the tree returned by these methods will be the true species tree). It is also known that concatenation (whether partitioned or unpartitioned) is not statistically consistent, and can even be positively misleading (i.e., converge to the wrong tree as the number of loci increases) (Roch and Steel, 2015; Roch et al., 2018). In general, establishing whether a method is statistically consistent or not is important for understanding its performance guarantees.
Yet, correspondingly little has been established about species tree estimation in the presence of GDL. For example, although likelihood-based approaches for species tree estimation have been developed [e.g., PHYLDOG (Boussau et al., 2013)], they have not been established to be statistically consistent. Key to understanding the performance of species tree estimation under GDL is whether the species tree topology itself is identifiable from the distribution it defines on the gene trees it generates. However, since gene trees can have multiple copies of each species when gene duplication occurs, this question can be formulated as: “Is the species tree identifiable from the distribution on MUL-trees?” where an MUL-tree is a tree with potentially multiple copies of each species.
In this article, we prove that unrooted species tree topologies are identifiable from the distribution implied on MUL-trees (Section 3) under the simple GDL model of Arvestad et al. (2009). Furthermore, we prove that the polynomial-time method ASTRAL-multi (Rabiee et al., 2019), a recent variant of ASTRAL designed to enable analyses of data sets with multiple individuals per species, is statistically consistent under this model (Section 3). We then present an experimental study evaluating ASTRAL-multi on 16-taxon data sets simulated under the DLCoal model (a unified model of GDL and ILS) (Rasmussen and Kellis, 2012); the results of this study show that when given a sufficiently large number of genes, ASTRAL-multi is competitive with other methods [e.g., DupTree (Bansal et al., 2010), MulRF (Chaudhary et al., 2014), and ASTRID-multi (Vachaspati and Warnow, 2015), the implementation of ASTRID for multiallele data sets] that also estimate species trees from MUL-trees (Section 4). We conclude with remarks about future work and implications for large-scale species tree estimation (Section 5).
Following the publication of the conference version of this work (Legried et al., 2020), identifiability of the species tree was extended by Markin and Eulenstein (2020); Hill et al. (2020) to the DLCoal (Rasmussen and Kellis, 2012), a unified model of GDL and ILS, and sample complexity results were obtained for ASTRAL/ONE under the DLCoal (Hill et al., 2020).
2. Species Tree Estimation from Gene Families
Our input is a collection
2.1. Astral
We provide theoretical guarantees and empirically validate an approach based on ASTRAL (Mirarab et al., 2014) in its variant for multiple alleles (Rabiee et al., 2019), which we refer to as ASTRAL-multi. Following Du et al. (2019), the input consists of unrooted MUL-trees
ASTRAL-multi proceeds as follows. Let S be the set of n species and let R be the set of m individuals. The input are the gene trees 
where
3. Theoretical Results
In this section, we provide theoretical guarantees for the reconstruction algorithm discussed in Section 2. Specifically, we establish statistical consistency under a standard model of GDL (Arvestad et al., 2009). First we show that the species tree is identifiable.
3.1. GDL model
We assume in this section that gene tree heterogeneity is due exclusively to GDL (and so no ILS) and that the true gene trees are known. That is, there is no gene tree estimation error (GTEE).
3.1.1. Birth/death process of GDL
The rooted n-species tree
3.2. Identifiability of the species tree under the GDL model
We first show that the unrooted species tree is identifiable from the distribution of MUL-trees
We begin with a quick proof sketch. The idea is to show that for each 4-tuple of species
It should be noted that the proof is not as straightforward as it is under the multispecies coalescent (Allman et al., 2011), as we explain next. Assume that the species tree restricted to
When all ancestral copies of
When the ancestors of a and b (or c and d) in R are the same, the species quartet topology results.
However, there are further cases. For example, if the ancestors of a and c in R coincide while being distinct from those of b and d, then the resulting quartet topology differs from that of the species tree.
Hence, one must carefully account for all possible cases to establish that the species quartet topology is indeed likeliest, which we do next. Our argument relies primarily on the symmetries (i.e., exchangeability) of the process.
Proof. It is known that the unrooted topology of a species tree is defined by its set of quartet trees (Bandelt and Dress, 1986). Let
In case 1, let R be the most recent common ancestor of
almost surely. Let 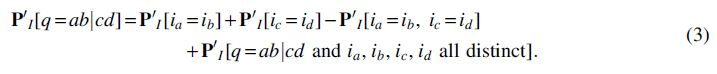
On the contrary,
and similarly for
where
For fixed y,
Proof. For 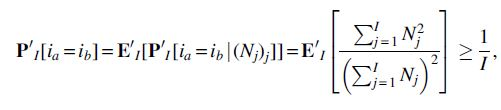
and similarly for
Returning to the proof of the theorem, evaluating h at
That establishes (2) in case 1, which implies
as desired.
The proof in case 2 is similar. Assume that
On the contrary,
with a similar result for
where
which establishes (2) in case 2. □
As a direct consequence of our identifiability proof, it is straightforward to establish the statistical consistency of the following pipeline, which we refer to as ASTRAL/ONE [see also Du et al. (2019)]: for each gene tree ti, pick in each species a random gene copy (if possible) and run ASTRAL on the resulting set of modified gene trees
Proof. First, we prove consistency for the exact version of ASTRAL. The input to the ASTRAL/ONE pipeline is the collection of gene trees 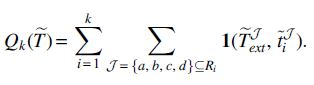
We note that the score only depends on the unrooted topology of
For a species 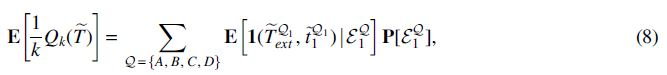
as, on the event  , there is no contribution from
, there is no contribution from
Based on the proof of Theorem 1, a different way to write
From (5), we know that this expression is maximized (strictly) at the true species tree
The default version is statistically consistent for the same reason as in the proof of Theorem 3. As the number of MUL-trees sampled tends to infinity, the true species tree will appear as one of the input gene trees almost surely. So ASTRAL returns the true species tree topology almost surely as the number of sampled MUL-trees increases. □
3.3. Statistical consistency of ASTRAL-multi under GDL
The following consistency result is not a direct consequence of our identifiability result, although the ideas used are similar.
Proof. First, we show that ASTRAL-multi is consistent when run in exact mode. The input are the gene trees 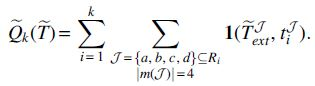
By independence of the gene trees (and non-negativity),
The expectation can be simplified as
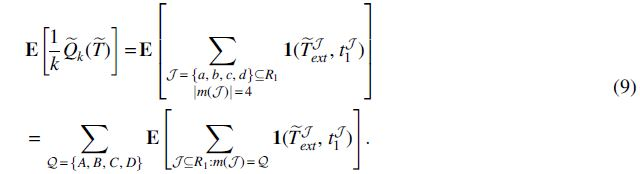
Let
when (without loss of generality)
It remains to establish (10). Fix
In case 1, let R be the most recent common ancestor of
which implies (10). By symmetry, we have
where here we define
Indeed, we then have:
Proof of Lemma 2. For 
where 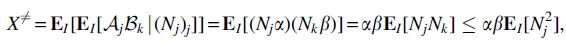
by Cauchy-Schwarz and
We now establish (11) in case 2. Assume that Contributions to Moreover, Arguing as in the proof of Theorem 1, by symmetry we have the equality For Similarly to case 1, by symmetry we have
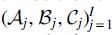 . Moreover, under PI,
. Moreover, under PI,  is independent of
is independent of  .
.
Putting these observations together, we obtain
where we used Lemma 2 on the last line.
Thus, ASTRAL-multi is statistically consistent when run in exact mode, because it is guaranteed to return the optimal tree, and that is realized by the species tree. To see why the default version of ASTRAL-multi is also statistically consistent, note that the true species tree will appear as one of the input gene trees, almost surely, as the number of MUL-trees sampled tends to infinity. For instance, the probability of observing no duplications or losses is strictly positive. Furthermore, when this happens, the true species tree bipartitions are all contained in the constraint set
4. Experiments
We performed a simulation study to evaluate ASTRAL-multi and other species tree estimation methods on 16-taxon data sets with model conditions characterized by three GDL rates, five levels of GTEE, and four numbers of genes. We briefly describe the study here and provide details sufficient to reproduce the study in Appendix A1. In addition, all scripts and data sets used in this study are available on the Illinois Data Bank: https://doi.org/10.13012/B2IDB-2626814_V1.
Our simulation protocol uses parameters estimated from the 16-taxon fungal data set studied by Du et al. (2019) and Rasmussen and Kellis (2012). First, we used the species tree and other parameters estimated from the fungal data set to simulate gene trees under the DLCoal (Rasmussen and Kellis, 2012) model with three GDL rates (the lowest rate
In our first experiment, we explored ASTRAL-multi on both true and estimated gene trees (Fig. 1). ASTRAL-multi was very accurate on true gene trees; even with just 25 true gene trees, the average species tree error was less than 1% for the two lower GDL rates and was less than 6% for the highest GDL rate (
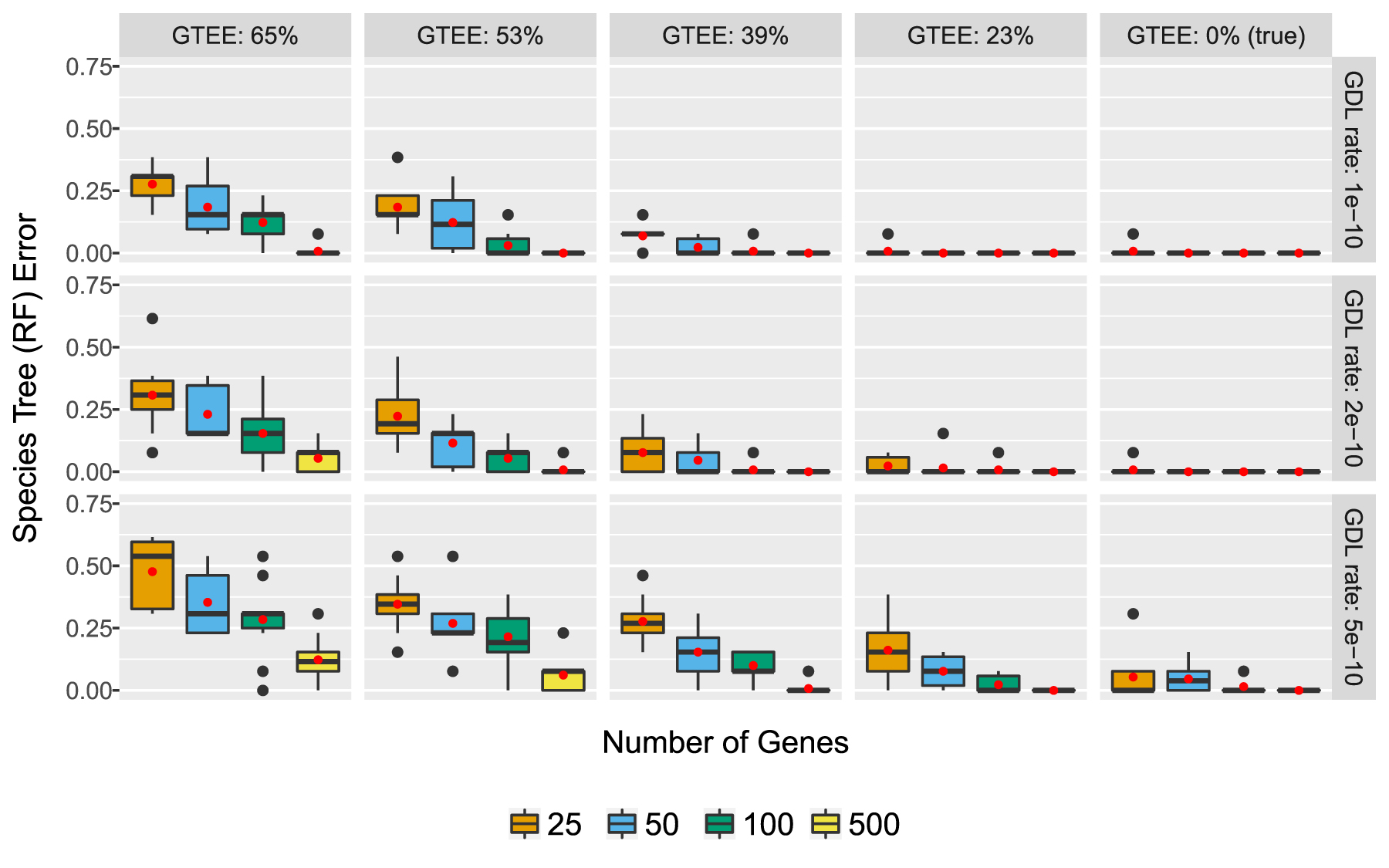
ASTRAL-multi on true and estimated gene trees generated from the fungal species tree (16 taxa) under a GDL model using three different rates (subplot rows). Estimated gene trees had four different levels of GTEE, by varying the sequence length (subplot columns). We report the average RF error rate between the true and estimated species trees. There are 10 replicate data sets per model condition. Red dots indicate means, and bars indicated medians. GDL, gene duplication and loss; GTEE, gene tree estimation error; RF, Robinson-Foulds.
In our second experiment, we compared ASTRAL-multi to four other species tree methods [DupTree (Wehe et al., 2008), MulRF (Chaudhary et al., 2014), STAG (Emms and Kelly, 2018), and ASTRID-multi, which is ASTRID (Vachaspati and Warnow, 2015) run under the multiallele setting] that take gene trees as input. Figure 2 shows species tree error for model conditions with mean GTEE of 53%. As expected, the error increased for all methods with the GDL rate and GTEE level, and decreased with the number of genes. Differences between methods depended on the model condition. When given 500 genes, all five methods were competitive (with a slight disadvantage to STAG); a similar trend was observed when methods were given 100 genes provided that the GDL rate was one of the two lower rates. When given 50 genes, ASTRAL-multi, MulRF, and ASTRID-multi were the best methods for the two lower GDL rates. On the remaining model conditions, ASTRID-multi was the best method. Finally, STAG was unable to run on some data sets when the GDL rate was high and the number of genes was low; this result was due to STAG failing when none of the input gene trees included at least one copy of every species. Results for other GTEE levels are provided in Tables 1, 2, and 3, and show similar trends.

Average RF tree error rates of species tree methods on estimated gene trees (mean GTEE: 53%) generated from the fungal 16-taxon species tree using three different GDL rates (subplot rows) and different numbers of genes (subplot columns). STAG failed to run on some replicate data sets for model conditions indicated by “NA,” because none of the input gene trees included at least one copy of every species.
For Each Model Condition (with Gene Duplication and Loss Rate
Species tree error was measured as the normalized RF distance between true and estimated species trees.
GDL, gene duplication and loss; GTEE, gene tree estimation error; RF, Robinson-Foulds.
For Each Model Condition (with Gene Duplication and Loss Rate
Species tree error was measured as the normalized RF distance between true and estimated species trees.
For Each Model Condition (with Gene Duplication and Loss Rate
Species tree error was measured as the normalized RF distance between true and estimated species trees. nan = NaN (Not a Number).
5. Discussion and Conclusion
This study establishes the identifiability of unrooted species trees under the simple model of GDL from Arvestad et al. (2009) and that ASTRAL-multi is statistically consistent under this model. In our simulation study, ASTRAL-multi was accurate under challenging model conditions, characterized by high GDL rates and high GTEE, provided that a sufficiently large number of genes is given as input. When the number of genes was smaller, ASTRID-multi often had an advantage over ASTRAL-multi and the other methods.
The results of this study can be compared with the previous study by Chaudhary et al. (2015), who also evaluated species tree estimation methods under model conditions with GDL. They found that MulRF and gene tree parsimony methods had better accuracy than NJst (Liu and Yu, 2011) (a method that is similar to ASTRID). Their study has an advantage over our study in that it explored larger data sets (up to 500 species); however, all genes in their study evolved under a strict molecular clock, and they did not evaluate ASTRAL-multi.
Our study is the first study to evaluate ASTRAL-multi on estimated gene trees, and we also explore model conditions with varying levels of GTEE. Evaluating methods under conditions with moderate to high GTEE is critical, as estimated gene trees from four recent studies (Jarvis et al., 2014; Hosner et al., 2016; Streicher et al., 2016; Blom et al., 2017) all had mean bootstrap support values below 50% [see table 1 in Molloy and Warnow (2018)], suggesting high GTEE.
Our study is limited to one underlying species tree topology with 16 species. Previous studies (Vachaspati and Warnow, 2016) have shown that MulRF (which uses a heuristic search strategy to find solutions to its NP-hard optimization problem) is much slower than ASTRAL on large data sets, suggesting that ASTRAL-multi may dominate MulRF as the number of species increases. Hence, future studies should investigate ASTRAL-multi and other methods under a broader range of conditions, including larger numbers of species. Future research should also consider empirical performance and statistical consistency under different causes of gene tree heterogeneity.
We note with interest that the proof that ASTRAL-multi is statistically consistent is based on the fact that the most probable unrooted gene tree on four leaves (according to two ways of defining it) under the GDL model is the true species tree (equivalently, there is no anomaly zone for the GDL model for unrooted four-leaf trees). This coincides with the reason ASTRAL is statistically consistent under the MSC as well as under a model for random horizontal gene transfer (HGT) (Roch and Snir, 2013; Daskalakis and Roch, 2016). Furthermore, previous studies have shown that ASTRAL has good accuracy in simulation studies where both ILS and HGT are present (Davidson et al., 2015). Hence ASTRAL, which was originally designed for species tree estimation in the presence of ILS, has good accuracy and theoretical guarantees under different sources of gene tree heterogeneity.
We also note the surprising accuracy of DupTree, MulRF, and ASTRID-multi, methods that, like ASTRAL-multi, are not based on likelihood under a GDL model. Therefore, DynaDup (Mirarab, 2019; Bayzid and Warnow, 2018) is also of potential interest, as it is similar to DupTree in seeking a tree that minimizes the duploss score (although the score is modified to reflect true biological loss), but has the potential to scale to larger data sets via its use of dynamic programming to solve the optimization problem in polynomial time within a constrained search space. In addition, future research should explore these methods compared with more computationally intensive methods such as InferNetwork_ML and InferNetwork_MPL [maximum likelihood and maximum pseudolikelihood methods in PhyloNet (Than et al., 2008; Wen et al., 2018)] restricted so that they produce trees rather than reticulate phylogenies, or PHYLDOG (Boussau et al., 2013) a likelihood-based method for coestimating gene trees and the species tree under a GDL model.
6. Appendices
6.1. Appendix A1: Details of simulation study
All scripts and data sets used in this study are available on the Illinois Data Bank: https://doi.org/10.13012/B2IDB-2626814_V1.
6.1.1. Simulation
Our simulation protocol is (largely) based on the 16-taxon fungal data set from Rasmussen and Kellis (2012).
6.1.1.1. Gene tree simulation
First, we used the species tree estimated by Rasmussen and Kellis (2012) (download: http://compbio.mit.edu/dlcoal/pub/config/fungi.stree) as the model species tree, modifying the branch lengths to be ultrametric and in generations, assuming 10 generations per year. Below is the Newick string for the resulting model species tree (height: 1,800,000,337.5 generations).
(((((((scer: 70617600.0, spar: 70617600.0): 49996800.0, smik: 120614400.0): 59706000.0, sbay: 180320400.0): 526823100.0, cgla: 707143500.0): 72206550.0, scas: 779350050.0): 231815475.0, ((agos: 785532600.0, klac: 785532600.0): 104349600.0, kwal: 889882200.0): 121283325.0): 788834812.5, (((calb: 412758000.0, ctro: 412758000.0): 296329500.0, (cpar:523231200.0, lelo: 523231200.0): 185856300.0): 311495850.0, ((cgui: 756158400.0, dhan: 756158400.0): 140068800.0, clus: 896227200.0): 124356150.0): 779416987.5);
We then simulated gene trees from this model species tree under the DLCoal model (Rasmussen and Kellis, 2012) (a unified model of GDL and ILS) with three different GDL rates using SimPhy Version 1.0.2 with the command:
simphy-1.0.2-mac64 -rs $nreps -rl F:ngens -rg 1 -s $stree \
-si F:1 -sp F:$psize -su F:$mrate -sg F:10 -lb F:$drate \
-ld F:lb -hg LN:1.5,1 -o <output directory> -ot 0 -om 1 \
-od 1 -op 1 -oc 1 -ol 1 -v 3 -cs 293745 &> <log file>
where $nreps is the number of replicate data sets (10), $ngens is the number of genes trees per data set (1000), $stree is the model species tree (Newick string above), $psize is the effective population size (
These parameters (with the GDL rate of
Fortunately, our parameter choice seems reasonable enough given that Rasmussen and Kellis (2012) considered values of 0.6 generations per year up to 10 generations per year for the fungal data set and selected
To understand the impact of our parameter choice, we simulated data sets assuming
(((((((scer: 7846400.0, spar: 7846400.0): 5555200.0, smik: 13401600.0): 6634000.0, sbay: 20035600.0): 58535900.0, cgla: 78571500.0): 8022950.0, scas: 86594450.0): 25757275.0, ((agos: 87281400.0, klac: 87281400.0): 11594400.0, kwal: 98875800.0): 13475925.0): 87648312.5, (((calb: 45862000.0, ctro: 45862000.0): 32925500.0, (cpar: 58136800.0, lelo: 58136800.0): 20650700.0): 34610650.0, ((cgui: 84017600.0, dhan: 84017600.0): 15563200.0, clus: 99580800.0): 13817350.0): 86601887.5);
From these simulations, we found that assuming
We Provide Summary Statistics for the Simulated Data Sets Below
All values shown below are averages (
We quantify the level of ILS as the normalized RF distance between each true locus tree and its respective true gene tree (which are on the same leaf set), averaged across all 1000 locus/gene trees (we refer to this value as the average distance or AD). Because these values are all less than 1% for the simulation assuming 10 generations per year, there is effectively no ILS in these data sets. The level of ILS is much higher for the simulation assuming
ILS, incomplete lineage sorting.
Last, unlike in the simulation study by Du et al. (2019), we did not enable gene conversion and allowed gene trees to deviate from the strict molecular clock by using the gene-by-lineage-specific rate heterogeneity modifiers (−hg). This means that a gamma distribution was defined for each gene tree by drawing
In summary, we used SimPhy to produce data sets with three model conditions, characterized by the three GDL rates; each of these model conditions had 10 replicate data sets, and each of these replicate data sets had 1000 gene trees.
6.1.1.2. Sequence simulation
For each model gene tree, we simulated a multiple sequence alignment (1000 base pairs) using INDELible Version 1.03 with GTR+GAMMA model parameters drawn from distributions; specifically, GTR base frequencies (A, C, G, T) were drawn from Dirichlet (113.48869, 69.02545, 78.66144, 99.83793), GTR substitution rates (AC, AG, AT, CG, CT, GT) were drawn from Dirichlet (12.776722, 20.869581, 5.647810, 9.863668, 30.679899, 3.199725), and
These distributions were based on the fungal data set from Rasmussen and Kellis (2012) (download: http://compbio.mit.edu/dlcoal/pub/data/real-fungi.tar.gz), which included a multiple sequence alignment estimated using MUSCLE (Edgar, 2004) and a maximum likelihood tree estimated using PhyML (Guindon et al., 2010) for each of the 5351 genes. We estimated GTR+GAMMA model parameters using RAxML Version 8.2.12 with the command:
raxmlHPC-SSE3 -m GTRGAMMA -f e -t <PhyML gene tree file> \
-s <MUSCLE alignment file> -n < output name>
We then fit distributions to the parameters estimated from alignments with at least 500 distinct alignment patterns and at most 25% gaps.
6.1.2. Gene tree estimation
On gene trees with four or more species, we estimated gene trees using RAxML Version 8.2.12 with the command:
raxmlHPC-SSE3 -m GTRGAMMA -p < random seed> -n < output name> \
-s <alignment file>
We truncated sequences to the first 25, 50, 100, and 250 base pairs to produce data sets with varying levels of GTEE. Sequence lengths of 25, 50, 100, and 250 resulted in mean GTEE of 65%, 53%, 39%, and 23%, respectively. Mean GTEE was measured as the normalized RF distance between true and estimated gene trees, averaged across all gene trees.
6.1.3. Species tree estimation
We estimated species trees using the first 25, 50, 100, or 500 (true or estimated) gene trees. ASTRAL Version 5.6.3 was run with the command:
java -Xms2000M -Xmx20000M -jar astral.5.6.3.jar -i <gene tree file> \
-a <name map file> -o <output file> &> <log file>
ASTRID Version 2.2.1 was run with the command:
/ASTRID -u -s -i <gene tree file> -a <name map file> \
-o <output file> &> <log file>
DupTree (download: http://genome.cs.iastate.edu/CBL/DupTree/linux-i386.tar.gz) was run with the command:
/duptree -i <gene tree file> -o <output file> &> <log file>
MulRF Version 2.1 was run with the command:
/MulRFSupertreeLin -i <gene tree file> -o <output file> &> <log file>
STAG (download: https://github.com/davidemms/STAG) was run with the command:
python stag.py <name map file> <gene tree folder> &> <log file>
We ran STAG with FastME Version 2.1.5 (Lefort et al., 2015). Importantly, STAG only uses gene trees that include at least one copy of every species. When the level of GDL was high (i.e.,
6.2. Appendix A2: Statistics on the number of copies per species in data sets
In Tables 5, 6, and 7, we report statistics on the number of copies per species in biological and simulated data sets.
For the Fungal Biological Data Set, We Report the Mean
For each species, we also report the fraction of gene trees with exactly one copy of the species as well as the fraction of gene trees with more than 1, 2, 5, 10, and 20 copies of the species (i.e.,
For a Data Set (Replicate 1) Simulated from the Estimated Fungal Species Tree Assuming 10 Generations per Year and a Duplication/Loss Rate of
For each species, we also report the fraction of gene trees with exactly one copy of the species as well as the fraction of gene trees with more than 1, 2, 5, 10, and 20 copies of the species (i.e.,
For a Data Set (Replicate 1) Simulated from the Estimated Fungal Species Tree Assuming
For each species, we also report the fraction of gene trees with exactly one copy of the species as well as the fraction of gene trees with more than 1, 2, 5, 10, and 20 copies of the species (i.e.,
Footnotes
Acknowledgments
All computational analyses were performed on the Illinois Campus Cluster and the Blue Waters supercomputer, computing resources that are operated and financially supported by UIUC in conjunction with the National Center for Supercomputing Applications.
Author Disclosure Statement
The authors declare they have no competing financial interests.
Funding Information
This study was supported, in part, by NSF grants CCF-1535977 and 1513629 (to T.W.) and by the Ira and Debra Cohen Graduate Fellowship in Computer Science (to E.K.M.). SR was supported by NSF grants DMS-1614242, CCF-1740707 (TRIPODS), and DMS-1902892, as well as a Simons Fellowship and a Vilas Associates Award. BL was supported by NSF grants DMS-1614242 (to S.R.) and CCF-1740707. Blue Waters is supported by the NSF (grants OCI-0725070 and ACI-1238993) and the state of Illinois.