
Abstract
Gene expression profiling makes it possible to conduct many biological studies in a variety of fields due to its thorough characterization of cellular states under various experimental conditions. Despite recent advances in high-throughput technology, profiling an entire set of genomes is still difficult and expensive. Due to the high correlation between expression patterns of different genes, the aforementioned problem can be solved with a cost-effective approach that collects only a small subset of genes, called landmark genes, representing the entire set of genes, and infer the remaining genes, called target genes, using a computational model. There are several shallow and deep regression models in literature to estimate the expressions of target genes from the landmark genes. However, the shallow mostly have limited capacity in learning the nonlinear and complex gene expression data and are prone to underfitting, and the deep models generally do not take advantage of correlation among target genes in the learning process and suffer from overfitting. Considering the gene expression inference as a multitask learning problem, we propose a new deep multitask learning algorithm to tackle these issues. Our learning framework automatically learns the correlation between target genes and uses this knowledge to improve its generalization. Specifically, we utilize a subnetwork with low-dimensional latent variables to discover the relationships between target genes and enforce a seamless and easy to implement regularization to our deep regression model. Unlike the existing multitask learning methods that can only deal with dozens or hundreds of tasks, our algorithm is able to efficiently learn the relationships between ∼10,000 target genes and, thus, is scalable to a large number of tasks. Our proposed method outperforms the shallow and deep regression models for gene expression inference and alternative multitask learning algorithms on two large-scale datasets regardless of the network architecture.
1. Introduction
A key problem in biological research is the characterization of cellular status in different states like disorders, drug therapeutics, and genetic perturbations. Gene expression profiling provides a powerful method for systematic cellular status analysis by identifying the patterns of gene expressions. Recent advances in high-throughput technologies allow comprehensive gene expression profiles to be collected under various cellular conditions, providing useful large-scale gene expression databases (Edgar et al., 2002; Brazma et al., 2003). For example, Van't Veer et al. (2002) recognized genes that are effective in breast cancer by investigating the patterns of gene expressions in various patients. By exploring the relations of gene expressions between different categories of tumors, Stephens et al. (2012) studied the relationships between and within various types of cancer. Richiardi et al. (2015) analyzed the gene expressions in a postmortem brain tissue and demonstrated the association between resting-state functional brain networks and gene activities, which are linked to ion channels and synaptic functions. A microarray study indicates a radical shift in the level of expression of several immune-related genes in mice susceptible to influenza A virus infection (Yan et al., 2015). In response to drug effects, gene expression patterns are also studied in various tasks such as construction of the drug-target network and drug discovery (Rees et al., 2016). Moreover, the connection of single-gene mutations on some chromosomes and early onset of Alzheimer disease are examined (Alzheimer's Association, 2013).
Despite recent progress on gene expression profiling, large archives of gene expressions are still costly and difficult to collect under various experimental conditions (Nelms et al., 2016). However, previous studies have shown that gene expressions are strongly correlated, suggesting that genes have similar functions in response to different conditions (Heimberg et al., 2016; Ntranos et al., 2016; Shah et al., 2016). The single cell RNA-Seq clustering studies also reveal a common pattern of expression between intracluster genes across various cell states (Ntranos et al., 2016). Therefore, the entire set of genes can be represented by a small subset of informative genes. This assumption was used by researchers in the Library of Integrated Network-based Cell-Signature (LINCS) program,* who used Principle Component Analysis to pick ∼1000 genes that contain ∼80% of the information of whole genome data. The cost of gene-expression profiling (∼$5 per profile) can be dramatically reduced by profiling these ∼1000 genes, named landmark genes, instead of the entire genomes (Peck et al., 2006). Therefore in profiling large-scale gene expression data, a cost-effective approach is to gather the landmark genes and estimate the remaining genes, named target genes, a computational model.
The first candidate for predicting target genes are the linear regression models with different regularizations. Some attempts were made to use nonlinear models to better capture the complex patterns of gene expression profiles guo2014inferring. In general, deep models have shown great versatility in learning the nonlinear patterns in biomedical data and high scalability when working with large databases. Inspired by the success of deep neural networks on several biological studies (Leung et al., 2014; Alipanahi et al., 2015; Spencer et al., 2015; Zhou and Troyanskaya, 2015; Singh et al., 2016), researchers introduced deep regression models for the task of gene expression inference (Chen et al., 2016; Ghasedi Dizaji et al., 2018). However, the interrelations between the target genes are not used by these deep regression models, which have several shared layers at the bottom and an exclusive layer at the top for different genes. In their training phase, these models consequently neglect the biological information associated with gene correlations, which contributes to their suboptimal results.
To tackle these challenges and benefit from the highly correlated target genes, we formulate the prediction of target genes from landmark genes as a multitask learning problem. In general, multitask learning algorithms seek to improve the generalization of predictors for multiple tasks using the information transferred through a joint learning system across similar tasks (Caruana, 1997). We consider each prediction of gene expression as a learning task and use the multitask learning model to capture the interrelationships between target genes as tasks to improve the prediction using this knowledge. Although there are several literature studies on the design of multitask learning algorithms for deep models (Ruder, 2017), they are mostly implemented on dozens or hundreds of tasks. Hence they are not efficient and scalable for gene expression inference with a large number of tasks (e.g., 10,000 tasks).
In this article, we propose a new multitask learning framework, denoted as Deep-LSMTL, for training deep regression networks that automatically learn the correlation between large number target genes (i.e., tasks) without high computational overheads. Deep-LSMTL particularly explores task relations by clustering of specific parameters of tasks. In other words, Deep-LSMTL aims to approximate reconstructing the parameters of each task by sparse combination of other parameters, providing a seamless regularization in stochastic training of deep models. Our algorithm uses a two-layer subnetwork with low-dimension bottleneck to capture the nonlinear low-rank representations of task relations. Furthermore, Deep-LSMTL alleviates the common issue of negative transfer in MTL methods by transferring asymmetric knowledge across the tasks and enforcing task correlation through the latent variables rather than the parameters. We evaluate Deep-LSMTL on two large-scale gene expression datasets compared to several deep and shallow regression models. Experimental outcomes show that our proposed algorithm has significantly better results in comparison to the state-of-the-art MTL methods and deep gene expression inference networks with different network size and architecture. In addition, we visualize the relevance of landmarks and target genes in the trained model to gain insight into gene relations. The main findings of this article can be summarized as the following points:
We introduce a new multitask deep regression method, which can address the large-scale prediction tasks and is effective for the gene expression inference problem, which has the nonimage data. We design a new seamless regularization for our deep multitask regression method to automatically learn the task interrelations by utilizing the multilayer subnetwork with low-rank latent variables. Our empirical studies show that the proposed new model can consistently outperform existing gene expression inference approaches and alternative multitask learning algorithms on two datasets regardless of network architectures.
The following sections in this study will be organized as follows. In Related Work section, we will briefly review the related works on recent multitask learning algorithms and gene expression inference task. In Deep Large-Scale Multitask Learning Network section, we will first revisit the general clustering-based multitask learning method and then introduce our new multitask deep regression model. After that, we will show the experimental results in Experiments section and evaluate the effectiveness of the proposed algorithm by comparing to other alternative models with different experimental conditions. In our empirical study, the visualization figures will also be plotted to validate the correctness of our method. Finally, we will conclude the article in Conclusion section.
2. Related Work
2.1. Gene expression inference problem
Finding a way to minimize the costs of gene expression profiling is an important issue in biological studies, since it is still difficult and expensive to archive whole-genome expression profiles under different perturbations and biological conditions (Nelms et al., 2016). Previous studies have shown that there is a strong correlation between gene expressions, and even a small set of genes can store extensive information. For example, Shah et al. (2016) showed that a random set of only 20 genes includes ∼50% of the information in the entire genes. In addition, recent RNA-seq studies support the idea that a small number of genes are adequate to store comprehensive information throughout the transcriptome (Heimberg et al., 2016; Ntranos et al., 2016).
Researchers from the LINCS program collected the GEO dataset † based on Affymetrix HGU133A microarrays and evaluated the similarity of gene expression profiles to determine the collection of most informative genes. They conducted experiments to calculate maximum percentage of information recovered by a subset of genes based on the comparable rank in the Kolmogorov–Smirnov statistic. Given the total number of 12,063 genes, they found that a subset of only 978 genes is sufficient to restore 82% of the observed connections in the whole transcriptome (Keenan et al., 2017). These genes (i.e., landmark genes) can be used to infer the expression of the other genes (i.e., target genes).
We can naturally formulate the gene expression inference problem as a multitask learning problem; some traditional models such as linear regression with
2.2. Multitask learning algorithms
The primary objective of multitask learning is to increase the generalization of several predictors in a shared training phase using the knowledge transferred through the associated tasks, while the MTL methods assume that the correlated tasks lie in a low-dimensional subspace (Caruana, 1997). Based on this hypothesis, Argyriou et al. (2008) applied
Sharing multiple layers across all tasks and stacking a particular layer for each task at the top is the standard way of adopting MTL methods on deep models. Furthermore, there are multiple works on designing the architecture of deep multitask networks (Yang and Hospedales, 2016, 2017; Ruder et al., 2017). To expand AMTL to deep neural networks, Deep-AMTFL transfers asymmetric task knowledge using latent variables instead of the model parameters (Lee et al., 2018). The proposed multitask learning algorithm is different with the previous approaches, because it provides an effective and scalable method for the gene expression problem with a large number of tasks using a bottleneck network for capturing the correlation between tasks.
3. Deep Large-Scale Multitask Learning Network
Considering
3.1. Clustered multitask learning
Multitask learning algorithms topically share the relevant knowledge between tasks using a joint learning framework for the tasks. This joint learning framework mostly includes a regularization term to improve generalization of the model like the following objective:
where
We can simply extend shallow MTL models to deeper MTL models by sharing a set of latent features across all tasks. Considering

The visualization of our Deep-LSMTL architecture.
One way for regularizing the task-specific parameters is to enforce clustering-based constraints based on the task correlations (Thrun and O'Sullivan, 1996; Bakker and Heskes, 2003; Evgeniou et al., 2005; Jacob et al., 2009; Lee et al., 2016). The clustering constraints force the correlated tasks to have similar parameters or features, but not all of the tasks to share features. This is helpful for avoiding the problem of negative transfer in MTL methods, where uncorrelated tasks degrade the shared features of related tasks (Ruder, 2017). A successful instance of the clustering constraints is using subspace clustering for grouping the task-specific parameters. Considering the subspace clustering constraint as the regularization in Eq. (2), we define the objective as follows:
where
We further multiply the features in the latest hidden layer to the parameters in the second term loss in Eq. (3). Then we are able to reformulate the objective in Eq. (3) like the following equation, as our last layer has the linear activation function:
where
3.2. Deep large-scale multitask learning
There are some disadvantages to the aforementioned model, such as lack of scalability to the large number of tasks like estimating ∼10,000 target genes from landmark genes), large number of parameters (i.e.,
To tackle these issues and efficiently learn the task correlations, we replace the linear function in
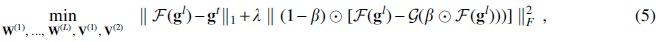
where
However, we cannot reconstruct the output of each task by the others in the 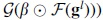 , and then only reconstruct the masked tasks according to the
, and then only reconstruct the masked tasks according to the
4. Experiments
In this section, we analyze our model in comparison with several shallow and deep regression models on two databases. In particular, we start by describing the different settings of the experiments, evaluating our proposed model compared to the state-of-the-art models through neural networks with various structures. We also try to shed light on learned knowledge stored in the model parameters by illustrating the connections of target and landmark genes.
4.1. Alternative methods
Least square regression mode is one of the most popular inference models. Its parameters are usually trained using this objective:
where
KNN is another well-known method for the regression problem, by which we obtain the output of test samples by averaging the k nearest samples in the training set. Furthermore, we include two deep learning methods, D-GEX (Chen et al., 2016) and SemiGAN (Ghasedi Dizaji et al., 2018), as the baseline. D-GEX model consists of a multilayer fully connected network, and SemiGAN adopts GANs in its regression model. However, our model utilizes a DenseNet architecture for its base network (i.e.,
In addition, we implement multiple MTL algorithms as the learning framework in the gene expression inference problem. Following, we briefly revisit them for our problem, but refer the readers to the original articles for more details. The CNMTL algorithm tries to group the specific parameters of tasks (i.e., last layer of the deep regression network in our case) through a regularization (Jacob et al., 2009). The following equation shows its objective, including the reconstruction loss and the regularization loss, based on the mean of parameters and the variances between and within clusters.
The second term shows the regularization term on the average of parameters with
The GO-MTL method enforces
where
The AMTL algorithm imposes a reconstruction loss on the task-specific parameters as the regularization as follows (Lee et al., 2018):
where
To expand AMTL to deep neural networks, Deep-AMTFL transfers related task knowledge using latent variable rather than the model parameters (Lee et al., 2016):
where
4.2. Datasets
We use three publicly available datasets as the data in our experiments, including the microarray-based GEO dataset, the RNA-Seq-based GTEx dataset, and the 1000 Genomes (1000G) RNA-Seq expression data. ‡ The GEO dataset contains 129,158 gene expression profiles corresponding to 22,268 genes, where the numbers of landmark and target genes are 978 and 21,290, respectively. These profiles are collected using the Affymetrix microarray platform in the RPKM format (i.e., Reads Per Kilobase per Million). The GTEx dataset consists of 2921 profiles, which are collected using the Illumina RNA-Seq platform. The 1000G dataset contains 2921 profiles from the Illumina RNA-Seq platform in the RPKM format.
We follow the preprocessing steps in Chen et al. (2016) in removing duplicate samples, joint quantile normalization, and cross-platform data matching. After removing the duplicated data, we map the expression values in the GTEx and 1000G datasets based on the quantile computed in the GEO data, resulting in the expression value in the range of 4.11 to 14.97. The expression values are then normalized to have zero mean and unit variance for each gene. Finally, there are 111,009 samples in GEO, 2921 samples in GTEx, and 462 samples in the 1000G datasets, where each sample (i.e., profile) includes 943 landmark genes and 9520 target genes.
We compare different methods under two settings as suggested in Chen et al. (2016). First, we consider 80% of the GEO data for training, 10% of the GEO data for validation, and the other 10% of the GEO data for testing. Second, we use the same 80% of the GEO data for training, the 1000G data for validation, and the GTEx data for testing. While the first case is a standard approach in selecting train, validation, and test sets, the second case is useful for validating the regression models on cross-platform prediction due to different distributions in train, validation, and test sets.
4.3. Evaluation metrics
We measure the effectiveness of inference models using two evaluation metrics, MAE and concordance correlation (CC). Denoting the predicted expressions as
where
where
4.4. Implementation details
We use DenseNet as the main architecture of our deep regression network with three 9000 dimensional hidden layers. We use Leaky rectified linear unit with leakiness hyperparameter
The Architecture of Deep-LSMTL Model
BN; FC; ReLU.
4.5. Performance comparison
In this subsection, we evaluate the performance of methods on predicting the target gene expressions on the two datasets. Figure 2 shows the results of different models in four groups, the shallow regression models in the first part, the existing deep regression networks in the second part, the MTL algorithms applied on deep regression models in the third part, and our DenseNet baseline and Deep-LSMTL network in the fourth part. We set the number of parameters for the deep MTL and our models based on the largest possible network fitted on GPU in this experiment. Consequently, Deep-Go-MTL, Deep-CNMTL, Deep-AMTFL, Deep-AMTL, and Deep-LSMTL have 8000, 4000, 5000, 7000, and 9000 hidden units, respectively.
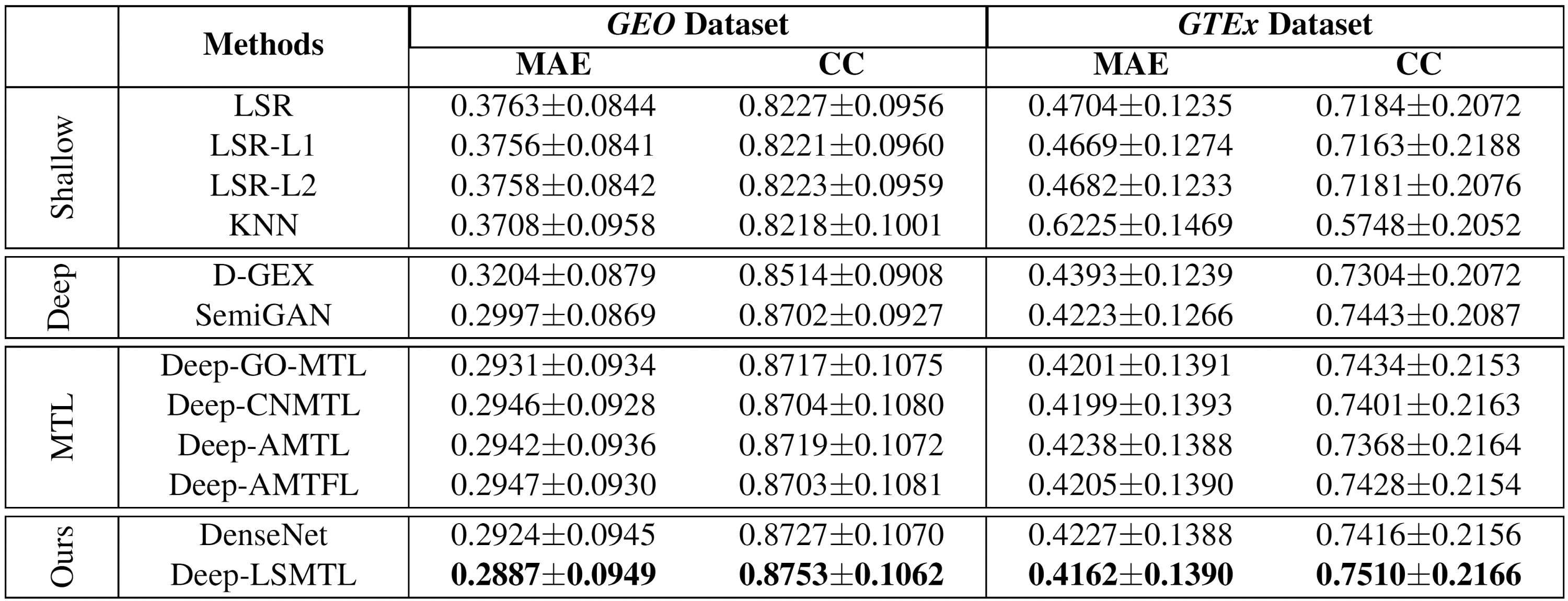
Experimental results to compare different machine learning methods on GEO and GTEx datasets based on the MAE and CC evaluation metrics. The empirical results of the traditional regression models are listed at the first part, and the previous deep inference networks are showed at the second part (these results were reported at their original articles or running their released codes). The results of multitask learning methods are reported at the third part, and the results of our proposed models are added at the fourth part using densely connected architecture with different numbers of hidden units. Better results correspond to lower MAE values or higher CC values. AMTL, asymmetric multitask learning; AMTFL; CC, concordance correlation; CNMTL; D-GEX; GAN, generative adversarial network; GEO; GO-MTL; GTEx; KNN, K-nearest neighbors; LSMTL; LSR; MAE, mean absolute error; MTL.
The first observation is that Deep-LSMTL has significant superiority compared to all of the other models consistently on both datasets according to both MAE and CC evaluation metrics. Deep-LSMTL substantially outperforms the shallow models as expected, showing the significance of deeper networks in learning the nonlinear gene expression data. Deep-LSMTL also provides better outcomes compared to the existing deep inference models in the literature, confirming the benefits of using the task interrelations in our multitask learning framework. Furthermore, Deep-LSMTL not only has better prediction in comparison with the alternative MTL algorithms but also it needs much less GPU memory than its MTL counterparts.
Since target gene expressions are normalized, it is probable that direct comparisons of the errors are not definitive. We use the
4.6. Ablation study
Although the previous experiments have validated the effectiveness of our model on large-scale multitask inference problems by providing a memory efficient network on one GPU, we develop a new experiment comparing our proposed algorithm and MTL methods on the networks with similar architecture. In particular, we set a two hidden-layer DenseNet as the architecture of all models in three different scenarios with 3000, 6000, and 9000 hidden units. Figure 3 indicates the results of Deep-GO-MTL, Deep-CNMTL, Deep-AMTL, Deep AMTFL, and Deep-LSMTL on both GEO and GTEx Datasets. It is worth mentioning that Deep-CNMTL and Deep-AMTFL still suffer from out-of-memory issues when they have 9000 hidden units. Figure 3 indicates that Deep-LSMTL consistently beats all the other MTL methods on various architectures. Therefore, our algorithm not only offers a better scalable method in training inference models with a large number of tasks but also demonstrates better outcomes in the case of identical base networks.
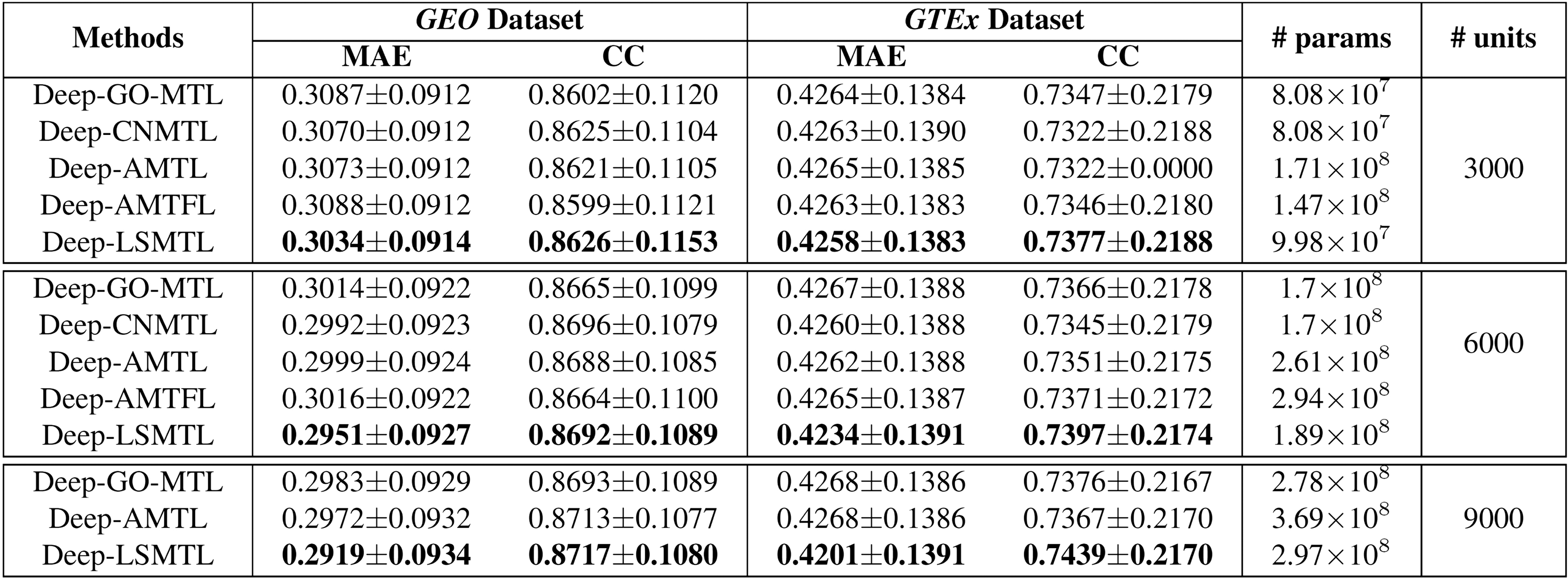
Experimental results to compare different deep multitask learning algorithms for the gene expression inference problems on GEO and GTEx datasets. All compared models use a two-hidden layer DenseNet as their structure, but have different numbers of hidden units at each part of the table. Better results correspond to lower MAE value or higher CC value.
In addition to examining the efficacy of Deep-LSMTL on base networks with DenseNet architecture, we analyze Deep-LSMTL and D-GEX with an MLP base network in Figure 4. The results are reported for both models with MLP base network containing one, two, or three hidden layers and 3000, 6000, or 9000 hidden units. Deep-LSMTL similarly outperforms D-GEX in all architectures consistently and confirms its capability irrespective of the base network choice.
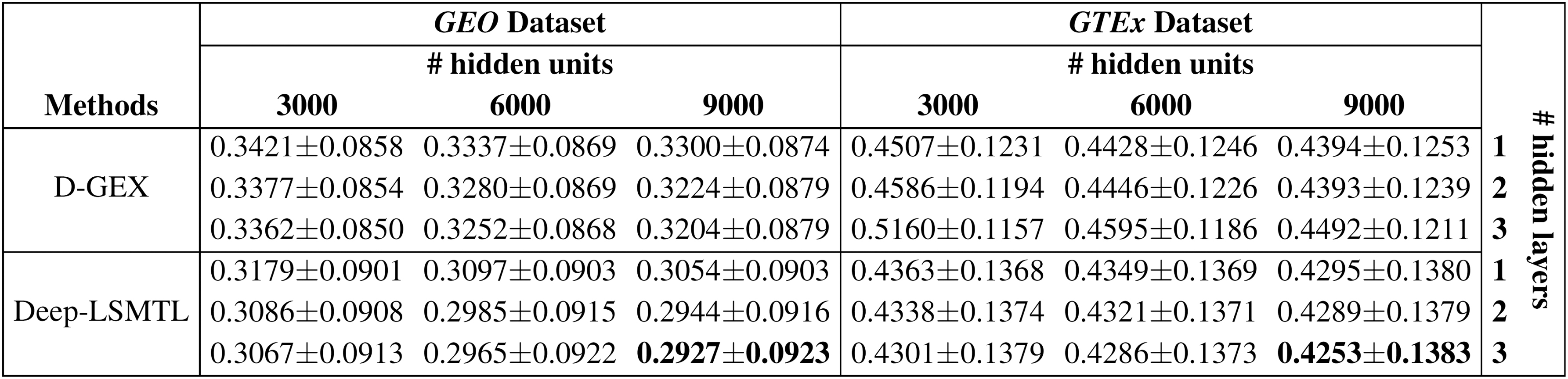
Experimental results to compare the MAE of D-GEX and Deep-LSMTL on GEO and GTEx datasets, when the number of hidden layers varies from 1 to 3 and the number of hidden units is 3000, 6000, or 9000. The structure of both models is based on the MLP network.
We also perform another ablation study to investigate the sensitivity of our learning framework with respect to the hyperparameters. Although we are able to select the dropout probability,
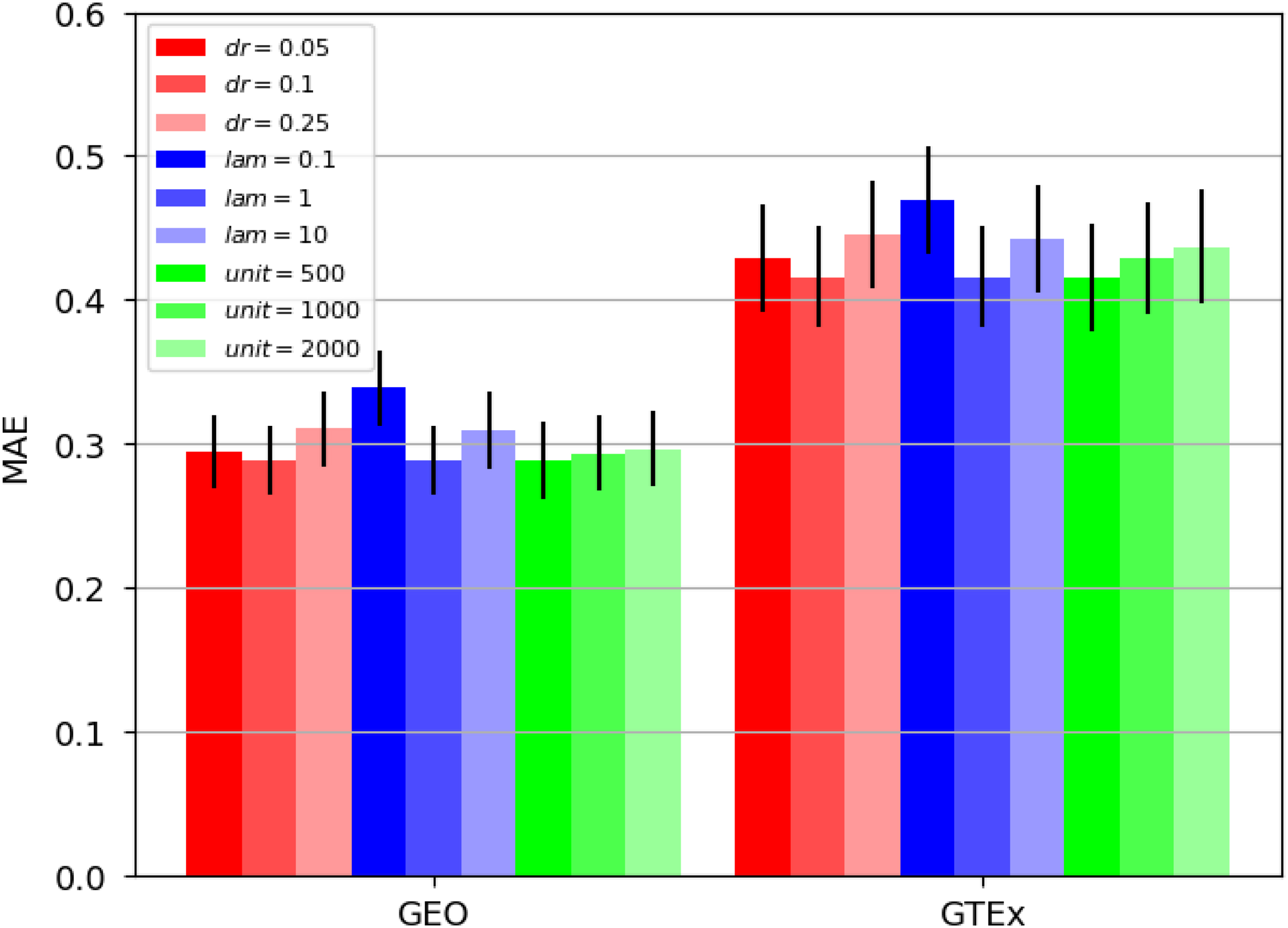
Ablation study of hyperparameter effects, including dropout probability (dr), the regularization hyperparameter
4.7. Visualization
To demonstrate the role of various landmark genes in our inference problem, we conduct a qualitative analysis on Deep-LSMTL by visualizing the role of different landmark genes in our trained model using the Layer-wise Relevance Propagation (Bach et al., 2015) technique. Figure 6 illustrates the results of Deep-LSMTL with DenseNet structure (in Table 1) on GEO dataset. Clustering the gene expression samples into 20 groups, Figure 6a and b shows the relevance score of landmark genes w.r.t. each cluster. The results suggest different patterns for different clusters of the landmark gene expressions, reproducing the findings in the earlier cancer subtype discovery and cancer landscape analysis that different groups of samples typically exhibit different expression patterns (Speicher and Pfeifer, 2015; Kandoth et al., 2013).
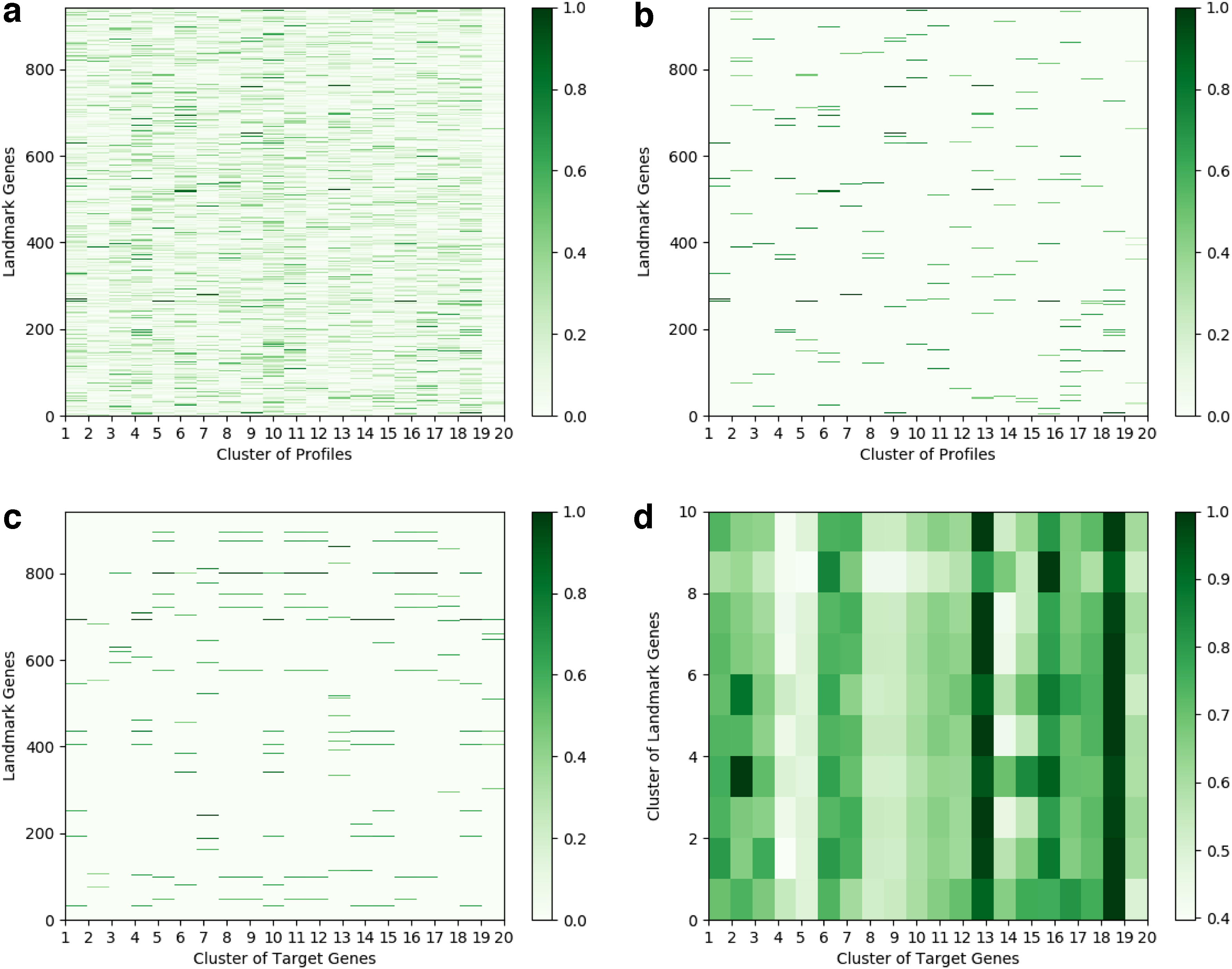
Visualization of the relevance score calculated for each landmark gene on GEO dataset.
We further explore the relationships between target and landmark genes. Dividing the target genes into 20 clusters, we calculate the overall relevance score of landmark genes in the prediction of each target gene cluster as shown in Figure 6c. Figure 6d provides better insights by categorizing the landmark genes to 10 groups and illustrating the connection between the clusters of target and landmark genes. While there are obvious disparities in the relevance patterns of different target gene clusters, there is correlation between some clusters. The previous gene cluster studies also showed similar findings on relations of gene clusters and the structure of biosynthetic pathways and metabolites (Medema et al., 2015).
We also visualize the predictions of our model on the GTEx dataset in Figure 7 similar to GEO dataset. The figures show similar patterns as the previous outcomes. However, they are more notable because of training on GEO data and predicting on GTEx data, qualitatively confirming the capability of our proposed model in capturing the relations among genes even for cross-platform prediction.
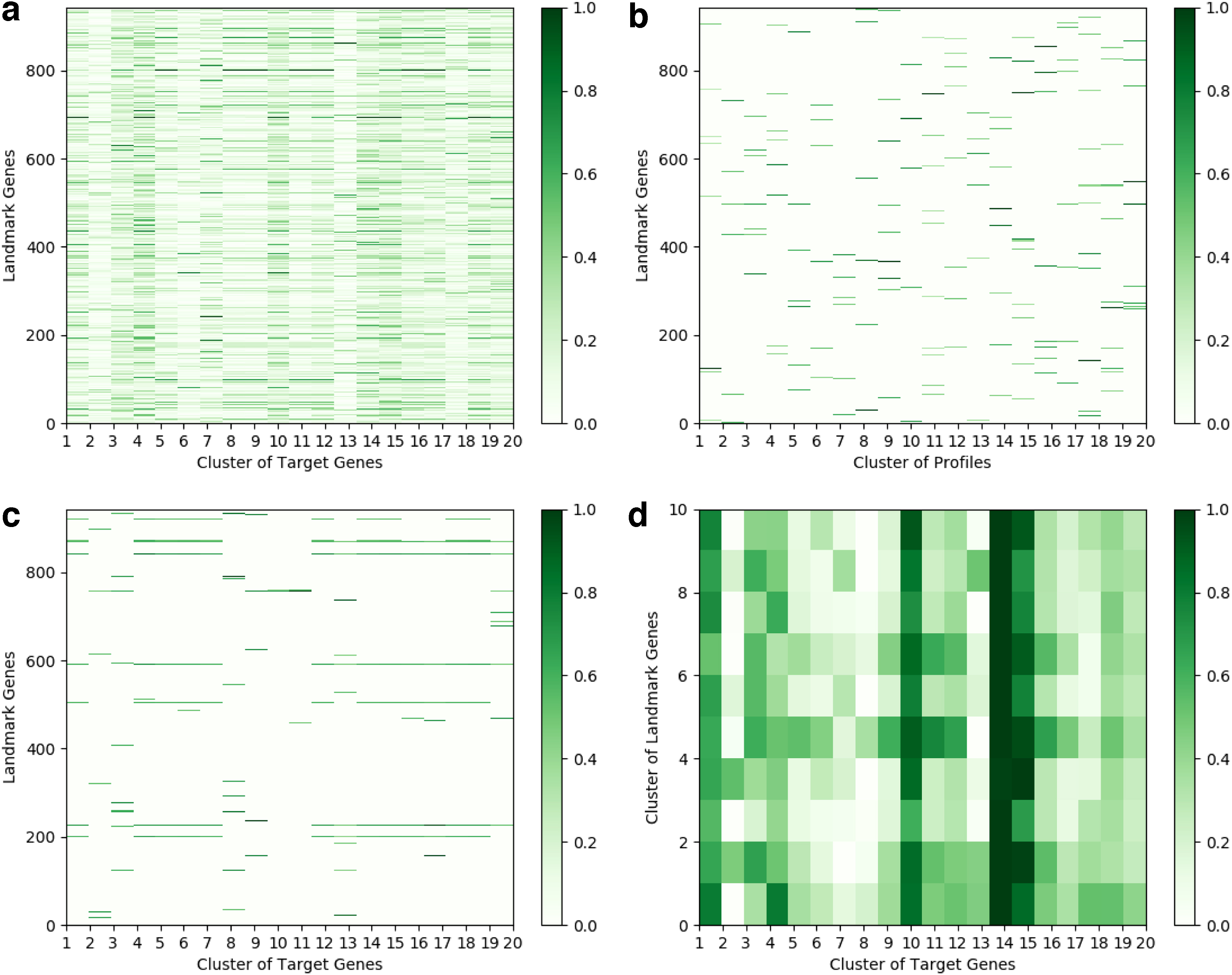
Visualization of the relevance score calculated for each landmark gene on GTX dataset.
5. Conclusion
In this study, we introduced a new MTL method for training deep inference models for estimating the gene expressions. Our algorithm improves the generalizations of multitask predictors by effectively discovering the task correlations. To do so, we proposed a seamless regularization for deep neural networks that is scalable to a huge number of tasks. Experimental results confirmed the effectiveness of our proposed algorithm compared to alternative models, where our model consistently and significantly outperforms all counterparts on two gene expression datasets with various base network architectures. We also visualized the role of landmark genes in estimating the expressions of target genes, providing better insights about the knowledge learned by our regression model.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was partially supported by NSF IIS 1845666, 1852606, 1838627, 1837956, 1956002, 2040588, and NIH AG049371.