
Abstract
While atom tracking with isotope-labeled compounds is an essential and sophisticated wet-lab tool to, for example, illuminate reaction mechanisms, there exists only a limited amount of formal methods to approach the problem. Specifically, when large (bio-)chemical networks are considered where reactions are stereospecific, rigorous techniques are inevitable. We present an approach using the right Cayley graph of a monoid to track atoms concurrently through sequences of reactions and predict their potential location in product molecules. This can not only be used to systematically build hypothesis or reject reaction mechanisms (we will use the ANRORC mechanism “Addition of the Nucleophile, Ring Opening, and Ring Closure” as an example) but also to infer naturally occurring subsystems of (bio-)chemical systems. Our results include the analysis of the carbon traces within the tricarboxylic acid cycle and infer subsystems based on projections of the right Cayley graph onto a set of relevant atoms.
1. Introduction
Traditionally, atom tracking is used in chemistry to understand the underlying reactions and interactions behind some chemical or biological system. In practice, atoms are usually tracked using isotopic labeling experiments. In a typical isotopic labeling experiment, one or several atoms of some educt molecule of the chemical system we wish to examine are replaced by an isotopic equivalent (e.g., 12C is replaced with 13C). These compounds are then introduced to the system of interest, and the resulting product compounds are examined, for example, by mass spectrometry (Chahrour et al., 2015) or nuclear magnetic resonance (Deev et al., 2019). By determining the positions of the isotopes in the product compounds, information about the underlying reactions might then be derived. From a theoretical perspective, characterizing a formal framework to track atoms through reactions is an important step to understand the possible behaviors of a chemical or biological system.
In this contribution, we introduce such a framework based on concepts rooted in semigroup theory. Semigroup theory can be used as a tool to analyze biological systems such as metabolic and gene regulatory networks (Egri-Nagy and Nehaniv, 2008; Nehaniv et al., 2015). In particular, Krohn–Rhodes theory (Rhodes et al., 2010) was used to analyze biological systems by decomposing a semigroup into simpler components. The networks are modeled as state automatas (or ensembles of automatas), and their characteristic semigroup, that is, the semigroup that characterizes the transition function of the automata (Mikolajczak, 1991), is then decomposed using Krohn–Rhodes decompositions or, if not computationally feasible, the holonomy decomposition variant (Egri-Nagy and Nehaniv, 2015). The result is a set of symmetric natural subsystems and an associated hierarchy between them, which can then be used to reason about the system. In Andersen et al. (2019), algebraic structures were employed for modeling atom tracking: graph transformation rules are iteratively applied to sets of undirected graphs (molecules) to generate the hyperedges (the chemical reactions) of a directed hypergraph (the chemical reactions network) (Andersen et al., 2013, 2016). A semigroup is defined by using the (partial) transformations that naturally arise from modeling chemical reactions as graph transformations. Utilizing this particular semigroup, the so-called pathway tables can be constructed, detailing the orbit of single atoms through different pathways to help with the design of isotopic labeling experiments.
In this work, we show that we can gain a deeper understanding of the analyzed system by considering how atoms move in relation to each other. To this end, we briefly introduce useful terminology in Section 2, found in graph transformation theory as well as semigroup theory. In Section 3, we show how the possible trajectories of a subset of atoms can be intuitively represented as the (right) Cayley graph (Dénes, 1966) of the associated semigroup of a chemical network. Moreover, we define natural subsystems of a chemical network in terms of reversible atom configurations and show how they naturally relate to the strongly connected components of the corresponding Cayley graph. We show the usefulness of our approach in Section 4.1 by using the constructions defined in Section 3 to differentiate chemical pathways, based on the atom trajectories derived from each pathway. We then show how the Cayley graph additionally provides a natural handle for the analysis of cyclic chemical systems such as the tricarboxylic acid (TCA) cycle (Harvey and Ferrier, 2010).
2. Preliminaries
2.1. Graphs
In this contribution, we consider directed as well as undirected connected graphs
Given two (un)directed graphs G and
Given a (directed) graph G, we call
Since the motivation of this work is rooted in chemistry, sometimes it is more natural to talk about the undirected labeled graphs as molecules, their vertices as atoms (with labels defining the atom type), and their edges as bonds (whose labels distinguish single, double, triple, and aromatic bonds, for instance), while still using common graph terminology for mathematical precision.
2.2. Graph transformations
As molecules are modeled as undirected labeled graphs, it is natural to think of chemical reactions as graph transformations, where a set of educt graphs are transformed into a set of product graphs. We model such transformations using the double pushout (DPO) approach. For a detailed overview of the DPO approach and its variations, see Habel et al. (2001). Here, we will use DPO as defined in the study of Andersen et al. (2016) that specifically describes how to model chemical reactions as rules in a DPO framework.
A rule p describing a transformation of a graph pattern L into a graph pattern R is denoted as a span
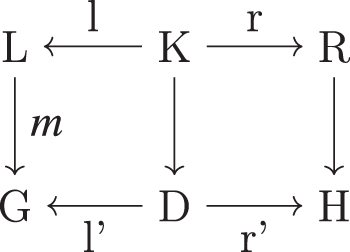
A direct derivation.
The graphs D and H are unique if they exist (Habel et al., 2001). The graph H is the resulting graph obtained by rewriting G with respect to the rule p. We call the application of p on G to obtain H via the map
For a DPO rule p to model chemistry, we follow the modeling in Andersen et al. (2013) and impose three additional conditions that p must satisfy. (1) All graph morphisms must be injective (i.e., they describe subgraph isomorphisms). (2) The restriction of graph morphisms l and r to the vertices must be bijective, ensuring that atoms are conserved through a reaction. (3) Changes in charges and edges (chemical bonds) must conserve the total number of electrons.
In the above framework, a chemical reaction is a direct derivation
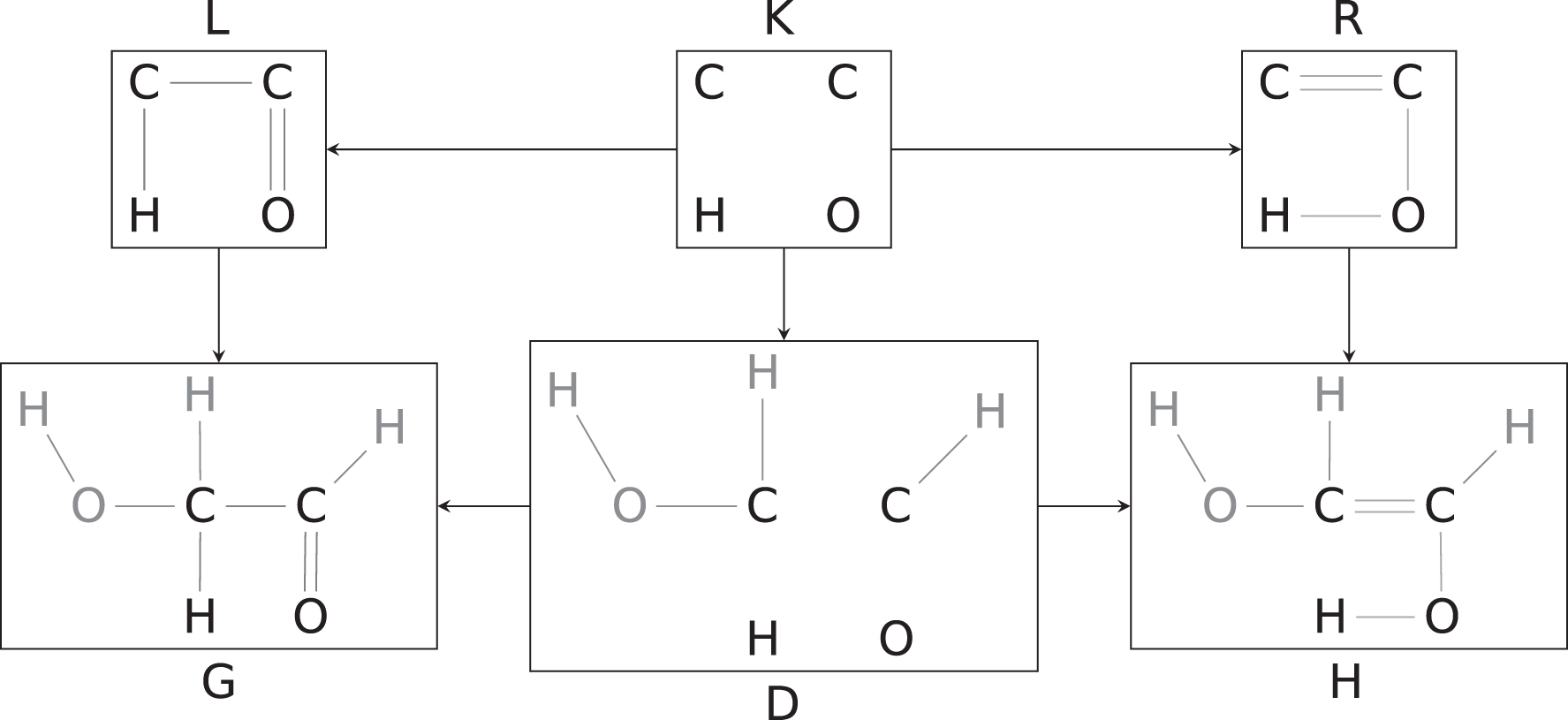
An example of a direct derivation. The mapping l, r,
2.3. Chemical networks
We consider a directed hypergraph where each edge
2.4. Semigroups and transformation semigroups
A semigroup is a pair
Given a nonempty finite set X, a transformation on X is an arbitrary map 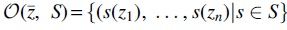 . In what follows, we use the notion
. In what follows, we use the notion
Given a transformation semigroup
3. Chemical Networks and Their Algebraic Structures
3.1. Characteristic monoids
Assume we are given some chemical network
The free monoid
Let
Often, we are only interested in tracking the movement of a small number of atoms. Let
We note that the above definitions are not unlike some of the core definitions within algebraic automata theory (Mikolajczak, 1991). Here, the possible inputs of an automata are often defined in terms of strings obtained from the free monoid on the alphabet of the automata. The characteristic semigroup is then defined as the semigroup that characterizes the possible state transitions. In the same vein, we can view our notion of event traces as the possible “inputs” to our chemical network CN that moves some initial configuration of atoms
In what follows, we let  ,
,  . Similarly, given a tuple of atoms
. Similarly, given a tuple of atoms 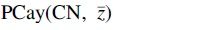 denote the projected Cayley graph
denote the projected Cayley graph
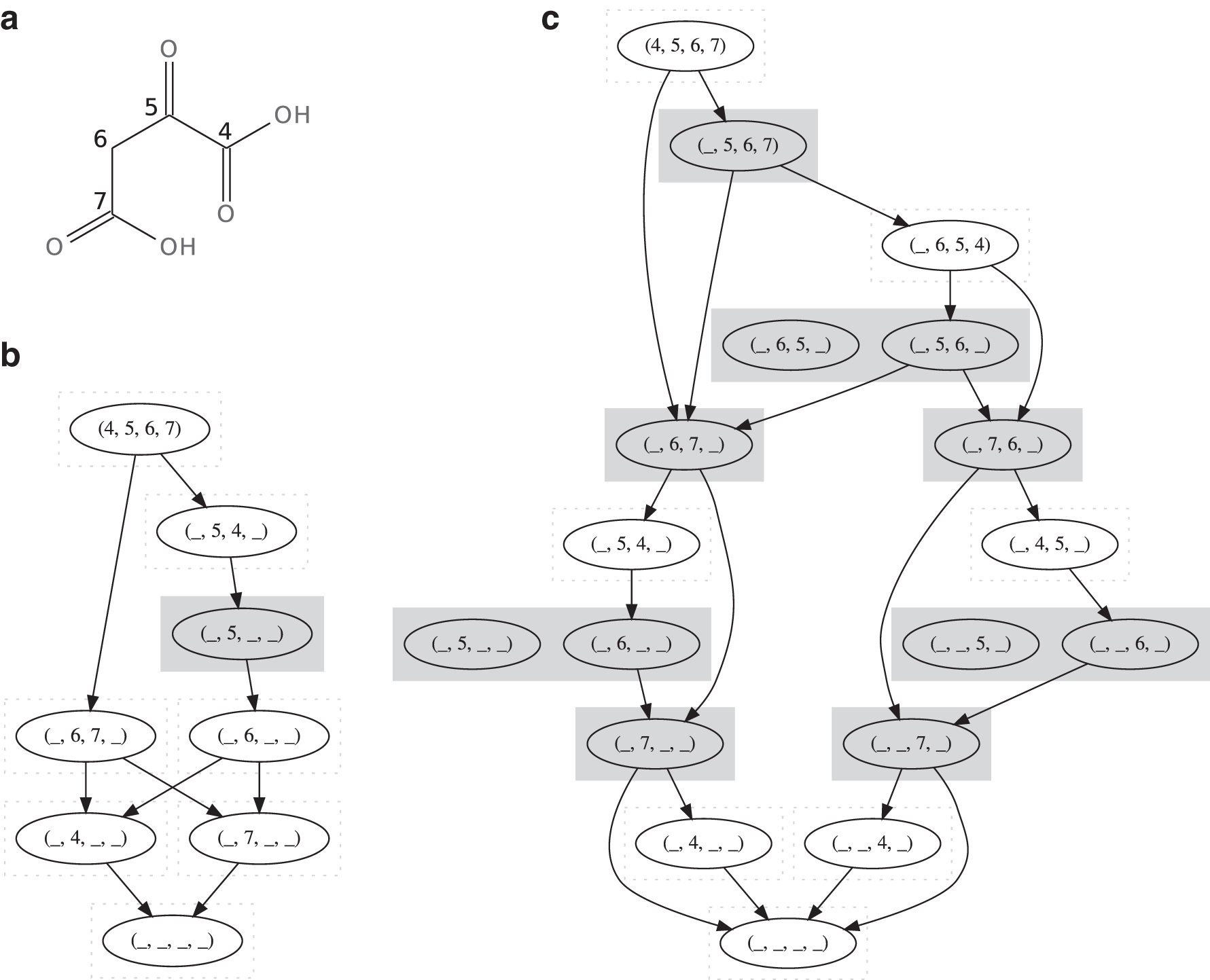
We can illustrate the relation between atom states using the Cayley graph

3.2. Natural subsystems of atom states
In the intersection between group theory and systems biology, attempts to formalize the notion of natural subsystems and hierarchical relations within such systems have been performed by works such as Nehaniv et al. (2015). Here, natural subsystems are defined as symmetric structures arising from a biological system. Such symmetries manifest as permutation groups of the associated semigroup representing said system. In such a model, the Krohn–Rhodes decomposition or the holonomy decomposition (Egri-Nagy and Nehaniv, 2015) can be used to construct a hierarchical structure on such natural subsystems of the biological system. In terms of atom tracking, however, defining natural subsystems in terms of the permutation groups in
The equivalence classes correspond to the strongly connected components of the Cayley graph
Notably, we observe from Figure 4b that the atoms 1 and 2 in the glycolaldehyde molecule can swap positions. We could of course also realize that such a swap was possible by noticing the symmetries in the glycolaldehyde molecule and the fact that we can convert glycolaldehyde to the
4. Results
4.1. Implementation
To test the practicality of the structures introduced in the previous section, we implemented the construction of the projected Cayley graph of a set of atoms in a chemical network. The resulting implementation can be found at https://github.com/Nojgaard/cat All code is written in python and uses the software package MØD (Andersen et al., 2016) and NetworkX (Hagberg et al., 2008) to construct the chemical networks and find the transformations used for the characteristic monoid. All figures in the following section were constructed with said implementation, and each run finished within seconds on an 8 core Intel Core i9 CPU with 64 GB memory. The most time-consuming part of the implementation was the computation of the transformations obtained from each hyperedge in the chemical network. In contrast, the construction time of the projected Cayley graph proved to be negligible.
4.2. Differentiating pathways
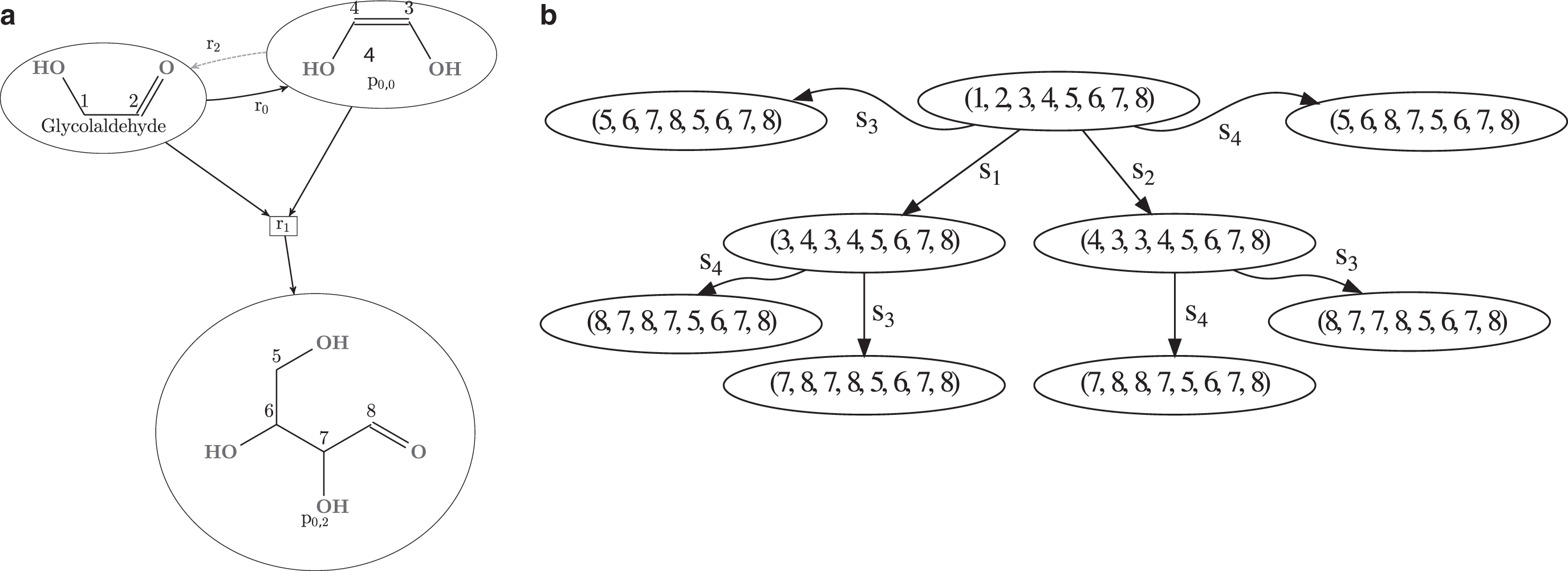
In this section, we will explore the possibilities of using the characteristic monoids of chemical networks to determine if it is possible to distinguish between two pathways P1 and P2, based on their atom states of their respective characteristic monoids. The motivation stems from methods such as isotope labeling. Here, a “labeled” atom is a detectable isotope whose position is known in some initial molecule and can then be detected, along with its exact position, in the product molecules of some pathway. In contrast to Andersen et al. (2019), we will not focus on the orbits of atoms in isolation, as we lose the ability to reason about atom positions in relation to each other. Moreover, as we will see here, the Cayley graph of the chemical network can be used to identify the exact event two pathways split.
Given a chemical network
Assume we wanted to device a strategy to decide what pathway is executed in reality. By replacing the nitrogen atoms of the E molecule with the isotope 13N, we would be able to observe where the atoms are positioned in the produced P molecule. Since we, by assumption, only label the nitrogen atoms of the E molecule, that is, the atoms 3 and 2, we can look at the orbits of the characteristic monoids
This fact also becomes immediately obvious by looking at the projected Cayley graph
4.3. Natural subsystems in the TCA cycle
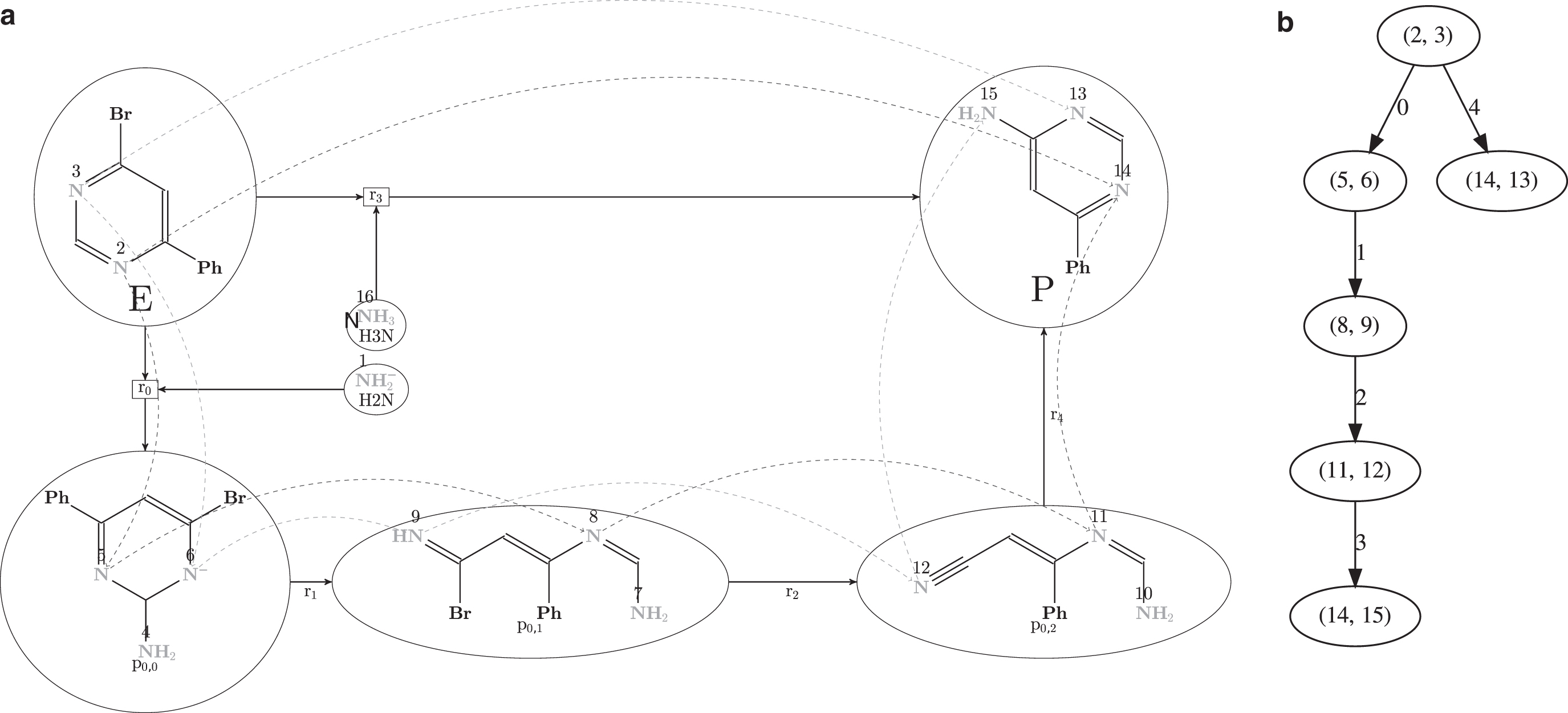
The citric acid cycle, also known as the TCA cycle or the Krebs cycle, is at the heart of many metabolic systems. The cycle is used by aerobic organisms to release stored energy in the form of ATP by the oxidation of acetyl-CoA into water and CO2. The details for the TCA cycle can be found in any standard chemistry text book, for example, Harvey and Ferrier (2010). In Smith and Morowitz (2016), the trajectories of different carbon atoms in the TCA cycle were examined to explain the change of their oxidation states. It is well known that there is an enzymatic differentiation of the two carboxymethyl groups in citrate, which requires a rigorous stereochemical modeling of the graph grammar rules used (Andersen et al., 2017). Ignoring such stereochemical modeling would lead to atom mappings not occurring in nature. We will provide a formal handle to analyze theoretically possible carbon trajectories using the algebraic constructs provided in this article. As we will see, such structures provide intuitive interpretations for the TCA cycle. More precisely, assume that we are interested in answering the following questions: What are the possible trajectories of the carbons of an oxaloacetate (OAA) molecule within the TCA cycle while (1) ignoring the enzymatic differentiation of the two carboxymethyl groups in citrate (denoted TCA- ) or (2) not ignoring (denoted TCA-
) or (2) not ignoring (denoted TCA- ). To answer these questions, we will decompose the characteristic monoid of the TCA cycle into its natural subsystems and examine them using the projected Cayley graph.
). To answer these questions, we will decompose the characteristic monoid of the TCA cycle into its natural subsystems and examine them using the projected Cayley graph.
In our setting, the TCA cycle is the chemical network and TCA- are modeled by the same network, the obtained transformations differ. More precisely, and .
A (simplified) chemical network modeling the TCA cycle. Note that any molecules not containing carbon atoms are modeled, but not depicted here. Each carbon atom is equipped with a unique ID for easy reference. TCA, tricarboxylic acid.
To start the cycle, an acetyl-CoA molecule is condensed with an OAA molecule, executing a cycle of reactions that ends up regenerating the OAA molecule while expelling two CO2 and water on the way. When an original atom is expelled from the cycle, we will consider it permanently lost. The carbon atoms of the OAA molecule that we are interested in tracking are annotated with the IDs 4, 5, 6, and 7. Let (resp. TCA-) consists of 213 (resp. 67) vertices. The full Cayley graphs are depicted in Figure 7a and b, respectively. When a carbon atom leaves the TCA cycle, we denote it by “_.” For example, the atom state
. The non-black-colored vertices of the same color correspond to atom states that are part of the same strongly connected component. 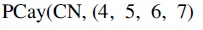 wrt. TCA-. The non-black-colored vertices of the same color correspond to atom states that are part of the same strongly connected component.
wrt. TCA-. The non-black-colored vertices of the same color correspond to atom states that are part of the same strongly connected component.
We can find the natural subsystems of (resp. TCA-), we find 92 (resp. 51) strongly connected components, of which 8 (resp. only 1) are nontrivial. Any nontrivial strongly connected component must invariably contain at least one tour around the TCA cycle since this is the only way the original atoms of the OAA molecules can be reused to create another OAA molecule. Moreover, any nontrivial strongly connected component represents a sequence(s) of reactions that uses (some of the) original atoms of the OAA molecule. To simplify and TCA- are depicted in Figure 8. Each box in the figure represents a natural subsystem that contains an atom state where every atom is either expelled or located in an OAA molecule. When ignoring the stereochemical formation of citrate, cycle turnover. However, in TCA-, only turnovers are executed. If that carbon changes location, it will leave the TCA cycle after exactly two more turnovers (the natural subsystems reachable from
Interestingly, . For example, the fact that the atom state , means that there exists a sequence of reactions that expels the carbons with IDs 4 and 7, but reuses the carbon atoms with IDs 5 and 6 to create a new OAA atom, where 5 is located at 6 and 6 is located at 7. Structurally, the atoms 4 and 7 correspond to the outer atoms in the carbon backbone in the OAA molecule, whereas the atoms 5 and 6 correspond to the inner atoms in the carbon backbone. In other words, the presence of
Figure 8c gives us a rough road map to determine exactly what sequence of events must have taken place to end up in the atom state
If any nontrivial strongly connected component in Figure 8c contains more than one vertex, it means that we can swap between atom states after a tour in the TCA cycle. As an example, consider the atom states corresponding to that natural subsystem of , as illustrated in Figure 9. The figure depicts all possible ways to swap the positions of atoms with IDs 5 and 6 as the possible paths between
The strongly connected component of containing the state
5. Conclusion
In this work, we have extended the insights provided by Andersen et al. (2019) by showing the natural relationship between event traces, the characteristic monoid, and its corresponding Cayley graph. The projected Cayley graph provides valuable insights into local substructures of reversible event traces.
We observe future steps for this approach to branch in at least two directions. On one hand, these methods show obvious applications in isotopic labeling design. To this end, it is natural to extend the system to model the actual process of such experiments. For example, when doing isotopic labeling experiments with mass spectrometry, molecules are broken into fragments and the weight of such fragments is deduced to determine the topology of the fragment. Using our model to track where the atoms might end up in such fragments and how it affects their weight seems like a natural next step. On the other hand, a more rigorous investigation of the fundamental properties derived from semigroup theory of the characteristic monoid seems appealing. As we have shown here, understanding such relations might grant insights into the nature of the examined system.
Footnotes
Author Disclosure Statement
The authors declare they have no competing financial interests.
Funding Information
This work was supported by Novo Nordisk Foundation grant NNF19OC0057834 and by the Independent Research Fund Denmark, Natural Sciences, grant DFF-0135-00420B.