
Abstract
For DNA sequence analysis, we are facing challenging tasks such as the identification of structural variants, sequencing repetitive regions, and phasing of alleles. Those challenging tasks suffer from the short length of sequencing reads, where each read may cover less than 2 single nucleotide polymorphism (SNP), or less than two occurrences of a repeated region. It is believed that long reads can help to solve those challenging tasks. In this study, we have designed new algorithms for mapping long reads to reference genomes. We have also designed efficient and effective heuristic algorithms for local alignments of long reads against the corresponding segments of the reference genome. To design the new mapping algorithm, we formulate the problem as the longest common subsequence with distance constraints. The local alignment heuristic algorithm is based on the idea of recursive alignment of k-mers, where the size of k differs in each round. We have implemented all the algorithms in C++ and produce a software package named mapAlign. Experiments on real data sets showed that the newly proposed approach can generate better alignments in terms of both identity and alignment scores for both Nanopore and single molecule real time sequencing (SMRT) data sets. For human individuals of both Nanopore and SMRT data sets, the new method can successfully math/align 91.53% and 85.36% of letters from reads to identical letters on reference genomes, respectively. In comparison, the best known method can only align 88.44% and 79.08% letters of reads for Nanopore and SMRT data sets, respectively. Our method is also faster than the best known method.
1. Introduction
Many fields in biological studies have been significantly changed by new sequencing technologies. For DNA sequence analysis, we are facing challenging tasks such as the identification of structural variants, sequencing repetitive regions, and phasing of alleles. Those challenging tasks suffer from the short length of sequencing reads, where each read may cover less than 2 single nucleotide polymorphism (SNP), or less than two occurrences of a repeated region. It is believed that long reads can help to solve those challenging tasks. However, the current long-read sequencing technology has high error rates. For DNA sequence analysis, the first step is read mapping. The other basic step is to align reads with the corresponding segments of reference genomes. There are lots of methods for read mapping and most of them do not work properly for long reads that have high error rates. A few new read mapping methods specifically for long reads are available now. However, they are still very slow. Besides, the accuracy of existing local alignment methods also needs to be improved when long reads are used.
The problem of read mapping has been well solved for short reads. A large number of tools have been developed for short reads that can map large-size data sets (such as data sets for human individuals) in reasonable time. It is known that Bwa might be the best tool for short reads (Li and Durbin, 2009). In general, tools for short reads cannot work well for large-size, long-read data sets. Recently, a few tools for long-read mapping have been developed. For single molecule real time sequencing (SMRT) data, Basic Local Alignment with Successive Refinement (Chaisson and Tesler, 2012) is a tool developed by the PacBio Company. In 2016, GraphMap (Sović et al., 2016) was proposed. It is the first tool that was reported to design for Nanopore data. The technique of minimizer was introduced in MashMap (Jain et al., 2017). Recently, Minimap (Li, 2016) and Minimap2 (Li, 2017) have been developed for long-read mapping. The most updated version Minimap2 can do read mapping and produce alignments of long reads against the corresponding segments in reference genomes. Minimap2 also uses the idea of minimizer to reduce the size of reference genomes and speed up the mapping algorithm. After that, a dynamic programming approach was designed to find the location of each read by calculating a score that represents the number of matched minimizers/k-mers and other facts.
In terms of local alignment, lots of tools have been developed for aligning reads with reference genomes. The seed-based method (Altschul et al., 1990) was first proposed in 1990. A seed DNA sequence is found based on a “hash table” containing all k-mers present in the first DNA sequence. The hash table is then used to locate the occurrences of the k-mer sequence in the other DNA sequence. Consequently, the seed is extended on both sides to complete the alignment. Other tools such as Blast (Kent, 2002), Soap (Li et al., 2008), SeqMap (Jiang and Wong, 2008), mrsFast (Hach et al., 2010), and Pass (Campagna et al., 2009) also used the seed technique. The method is simple and quick for shorter sequences. However, it is more memory-intensive for long sequences.
PatternHunter (Ma et al., 2002) extends the seed approach and proposed the “spaced seeds” concept. The main idea is to require only some positions of the seed to be matched. Some methods using multiple seed hits and allowing indels include Shrimp (Rumble et al., 2009) and RazerS (Weese et al., 2009). Several very fast tools such as Ssahs2 (Ning et al., 2001), Bwasw (Li and Durbin, 2010), Yoabs (Galinsky, 2012), and BowTie (Langmead et al., 2009) have been created based on the “Burrows-Wheeler Transform,” a technique that was first used for data compression (Burrows and Wheeler, 1994). Alignment tools such as Soap3 (Liu et al., 2012a), BarraCUDA (Klus et al., 2012), and Cushaw (Liu et al., 2012b) combine “retrieval-based” approaches with general-purpose computing on graphics processing units computing, taking advantage of parallel graphics processing unit (GPU) cores to accelerate the process. Nevertheless, the above approaches are still very slow when handling Nanopore and SMRT data sets.
Single instruction multiple data (SIMD) is a technology that uses a single instruction to control multiple processing units and perform the same operation on each set of data (also known as “data vector”) to achieve data-level parallelism. This technique is used for accelerating the dynamic programming-based pairwise alignment algorithms (Needleman and Wunsch, 1970; Smith and Waterman, 1981). Farrar's striped dynamic programming algorithm (Farrar, 2007) is the most popular one for applying SIMD technique in pairwise alignment. Ssw (Zhao et al., 2013) and Parasail (Daily, 2016) both adopted the Farrar's algorithm. Suzuki and Kasahara (2018) improved Farrar's algorithm by introducing a difference recurrence relation. Minimap2 adopted Suzuki and Kasahara's algorithms for fast pairwise alignment between the fixed anchors. SIMD technique is a key for the success of Minimap2. The speed of Minimap2 is very fast. On multi- and many-core processors, the combined SIMD vectorizing strategies are adopted for hardware accelerating of pairwise alignment algorithms (Hou et al., 2016), proposing an efficient vectorizing framework. GPU and Field Programmable Gate Array are also used for hardware accelerating of pairwise alignment algorithms. Some methods using GPUs are given in Striemer and Akoglu (2009), Benkrid et al. (2012), Li et al. (2014), Ahmed et al. (2019), and Sundfeld et al. (2020).
In this study, we have designed new algorithms for mapping long reads to reference genomes. We have also designed efficient and effective heuristic algorithms for local alignments of long reads against the corresponding segments of reference genome. To design the new mapping algorithm, we formulate the problem as the longest common subsequence with distance constraints (LCSDCs). The local alignment heuristic algorithm is based on the idea of recursive alignment of k-mers, where the size of k differs in each round. We have implemented all the algorithms in C++ and produce a software package named mapAlign. Experiments on real data sets showed that the newly proposed approach can generate better alignments in terms of both identity and alignment scores for both Nanopore and SMRT data sets.
For human individuals of both Nanopore and SMRT data sets, the new method can successfully math/align 91.53% and 85.36% of letters from reads to identical letters on reference genomes, respectively. In comparison, the best known method can only align 88.44% and 79.08% letters of reads for Nanopore and SMRT data sets, respectively. Our method is also faster than the best method. Here we did not use the SIMD technique as in the state-of-the-art method. Our new mapping method is based on the model, the longest common subsequence with approximate distance constraint (
2. Methods
The whole method includes two parts, where the first part identifies the location of each read in the reference genome and the second part aligns the read against the corresponding segment in the reference genome. The second part is referred to as local alignment.
2.1. Identifying the location of a read in the reference genome
A common technique for all the existing methods for identifying the location of a long read in the reference genome is to start with a set of k-mers from the read and based on the set of k-mer positions in the reference genome to find the correct location of the read in the reference genome. The reason to use multiple k-mers is due to the fact that the length of long read could be very long, for example, 100k base pairs. Various methods use various strategies on selecting multiple k-mers.
2.1.1. Reference genome index
Homopolymer-compressed (HPC) k-mers are used to improve overlap sensitivity for SMRT reads (Li, 2017). It was first proposed by SmartDenovo (https://github.com/ruanjue/smartdenovo; J. Ruan). An HPC k-mer is obtained from a sequence by compressing every subsequence of identical letters into one letter. It contains k letters where any two consecutive letters in the k-mer are different. We use HPC k-mers for reference genomes.
The popular approach to convert a k-mer to an integer index is 4-based hush function in Eq. (1). It convert a k-mer string
To increase the speed of our algorithm, here we use another approach. We directly use an array of size
The compressed k-mer and new hush function have the following advantages. (1) We can noticeably reduce the key space of hash function for converting a k-mer, for example, the keys spaces of a k = 15 k-mer are
The Distribution of Original k-mer Sequence Length of Compressed k-mer for k = 14, 15, and 16 (Chromosome 1 of GRCh38)
2.1.2. Identifying the location of a read
To find the location of a read in the reference genome, the main difficulty is that each k-mer on a read may appear at several places in the reference genome. To find the “true” location of a read in the reference genome, multiple k-mers on the read should be used. To handle multiple k-mers, we need to design corresponding efficient and effective algorithms.
2.1.2.1. Sampling k-mers from a read
There are different ways to sample k-mers. The first issue is how many k-mers to sample. The other issue is how to combine those k-mers’ information to identify the true location of the read in the reference genome. GraphMap needs to use all the k-mers on a read and decomposes the whole genome into a set of buckets (with overlap) and looks at the number of k-mers in each bucket (Sović et al., 2016). Note that, the number of buckets is proportional to the total length of the genome; the number of buckets may be very large for large genomes. Moreover, the number of k-mers in each bucket is not an accurate measure, where the order among the k-mers and the distance between two consecutive k-mers are not taken into consideration. Thus, they have to use all the k-mers on the read in the process. Therefore, the running time is very slow compared with the best known tools, for example, Minimap2 (Li, 2017).
To reduce the number of k-mers, Minimap2 adopted the technique of minimizer and designed a dynamic programming heuristic approach to identify the true location of read in the reference genome, where an objective function considering the number of matched k-mers and other facts was used.
In this study, we propose a new method that needs a relatively small number n of k-mers approximately evenly distributed over the read. Here the default value is
For a read r, we select n k-mers that are evenly distributed over the read. Let
Each k-mer ri occurs at several locations over the reference genome. Let
2.1.2.2. The LCSDC problem
Let
Since there are indels, we have the approximate version, the
2.1.2.3. An
expected time exact algorithm for LCSDC
Let
It is easy to see that the running time of Algorithm 1 is linear in terms of the total length of Li and
The complete algorithm is given as follows. We merge Li and
It is easy to see that the merge process only works well for the first round and fails on the rest of the rounds. The merge process heavily depends on the fact that the “distance” between the two lists is a fixed value d. For the newly created lists, there are items from different lists. Thus, the distance between two items from the two merged lists may differ. For example, the distances between two items from
To overcome the problem, we need to move each item y in
Proof. The first step is to move every item in list Li for
2.1.2.4. An
running time heuristic algorithm for
When there are indels, the approximate version
First, we remove identical k-mers within distance
2.1.2.5. Shifting
Note that the running time is proportional to the total length of the k-mer lists. To reduce the running time, for each of the n k-mers from the read, we look at the next m base pairs with sliding step s and choose the one with the shortest list. Since the read length may vary from hundreds to millions of base pairs, we use a shifting stride to adjust m and s according to the read length l and k-mer number n. The shifting strides are given in Table 2.
The Shifting Stride for Different Read Lengths
2.1.2.6. Implementation of the algorithms
We have implemented the algorithm in C++. For each k-mer, the list containing all occurrences of the k-mer in genome is actually stored in an array (instead of a linked list) and this is one of the keys to speed up the program. It is perhaps worth emphasizing that accessing a list for a k-mer in a huge size array/hash table is time-consuming, and we access the huge size array/hash table only once for each k-mer on the read. When the final list is obtained after
We set the k-mer number
We use small-size Nanopore and SMRT data sets (see Section 3.1, data set 3 and data set 4) to test the tool for the
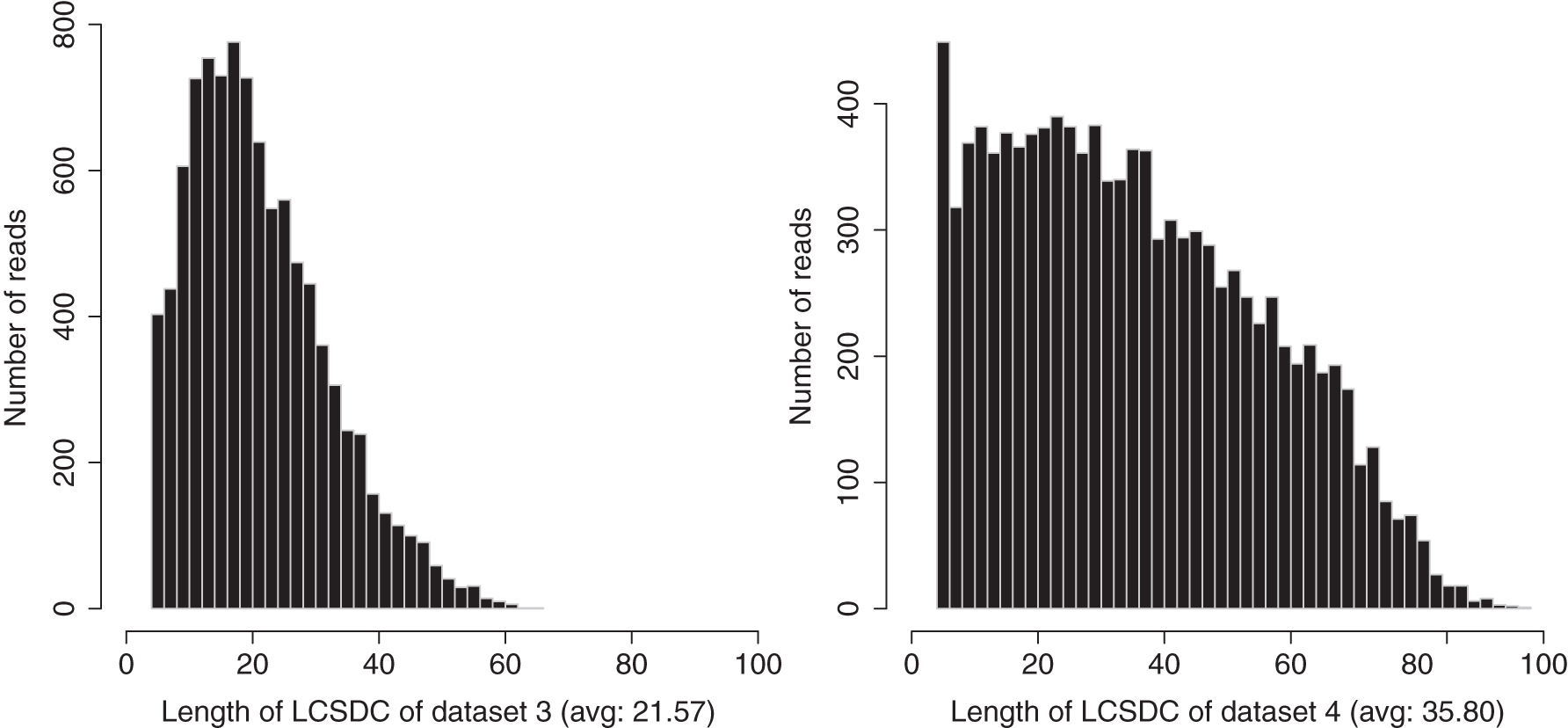
The distributions of the LCSDC lengths for the data set 3 (Nanopore) and data set 4 (SMRT) with GRCh38 as the reference. LCSDC, longest common subsequence with distance constraint; SMRT, single molecule real time sequencing.
2.1.2.7. k-mer size versus the algorithm speed
The running time of our read mapping algorithm is
To illustrate the speed of our algorithm for different k values, we did experiments on a Nanopore data set and an SMRT data set (see Section 3.1, data set 3 and data set 4). The results are given in Table 3. We can see that when k increases, the running time decreases and the average length of LCSDC also decreases slightly. For
The Comparison of Our Algorithm for Data Set 3 and Data Set 4 with Different k-Values of k-mer (GRCh38 As the Reference)
2.1.2.8.
value versus the LCSDC length
The
The Longest Common Subsequence with Distance Constraint Length of Our Algorithm for Data Set 3 and Data Set 4 with Different r-Values (GRCh38 As the Reference and k-mer Size = 15)
2.2. Algorithms for local alignment
Once the read location on reference genome is found, we need to align the read with the reference genome. Of course, a quadratic running time dynamic programming algorithm can give the optimum solution. However, the algorithm is too slow to handle large-size data sets such as Nanopore or SMRT data for human individuals. Here we propose a heuristic algorithm with running time proportional to the length of the sequences for local alignment. Moreover, our method is much more accurate than all the existing heuristic methods.
We treat the DNA sequences (with 4 letters) as a sequence of k-mers. When the DNA sequence is long, we use a large-size k-mer to find an alignment of the k-mers between the two sequences. Such an alignment of k-mers between the two sequences decomposes the whole sequence region into many smaller size regions and for each pair of smaller size regions we can recursively use smaller size k-mers to do the alignment. When the size of the pair of regions is small enough, for example,
Recall that our program has found an LCSDC for k-mers with
2.2.1. The heuristic algorithm for alignment of k-mer sequences
Let
It is easy to see that the running time of Algorithm 3 is linear in terms of the input sequences and is much simpler and faster than the heuristic algorithm for
Next, we go to the last pair of matched k-mers (with
If the returned
3. Results
In this section, we compare mapAlign with different existing methods in terms of the running time and alignment quality. We use some public data sets to illustrate.
3.1. Data sets
Two large human data sets are used for full comparison purpose (data set 1 and data set 2). We also extracted two downsized data sets (data set 5 and data set 6) of different human individuals from National Center for Biotechnology Information for further comparison. Two small-size human data sets (data set 3 and data set 4) are generated for quick testing. A detailed description of these data sets is shown in Table 5. Human genome GRCh38 is used as the reference genome. Experiments were conducted under the Ubuntu 16.04 operating system and a cluster with 96 CPUs (Intel(R) Xeon(R) CPU E7-8860 v3 @ 2.20GHz).
The Detailed Description of the Data Sets Used for Experiments
SMRT, single molecule real time sequencing.
3.2. Alignment quality comparison with Basic Local Alignment Search Tool
We define two different scores (alignment score and identity score) to evaluate the quality of the generated alignments. The alignment score is defined as the number of identically matched pairs of letters over the total length of read. The identity score is defined to be the number of identically matched pairs of letters over the total length of the alignment. The alignment score quantifies the percentage of matched read letters, while the identity score provides the percentage of identically matched columns over the alignment. If we align a read to itself, both the alignment score and identity score should be 100%. A reasonable alignment approach should try to achieve high values for both alignment score and identity score. Using one either the alignment score or identity score may be a bias. For example, all letters of a read with 200 base pairs may be perfectly matched to a long segment of 2000 base pairs in the reference genome. Such an alignment has high alignment score and low identity score, and should be considered very poor. On the contrary, a read with 2000 letters may have 200 consecutive letters that are identical to a segment of 200 letters in a reference genome. Then we will have a local alignment containing 200 identical alignments. The identity score is 100% in this case, while the alignment score is 10%, that is, only 10% of letters in the read are matched.
The Basic Local Alignment Search Tool (BLAST; Altschul et al., 1990) is good at doing alignment. To evaluate the performance of our method, we did a preliminary comparison with BLAST (version 2.9.0). To accelerate the experiments, we use a small human data set (data set 3) against the human reference GRCh38. The experiment results are as follows. The alignment score and identity score of mapAlign are 89.70% and 75.80%, while the alignment score and identity score for BLAST are 70.40% and 77.77%, respectively. Our method has a slightly lower identity score than BLAST. However, our method achieved better alignment score. A total of 89.70% of read letters are perfectly matched to the reference genome by our method, and BLAST could only align 70.40% of the read letters. We carefully compared the details of the alignments generated by our method with that of BLAST. We find that our method aligned slightly more space letters to the reference genome to get a better alignment score. Extra
3.3. Comparison with other methods
According to Li (2017), Minimap2 is over 30 times faster than GraphMap. Minimap2 also has better alignment quality than GraphMap. Here we compare mapAlign with Minimap2 using two large-size data sets, that is, data set 1 and data set 2. The results are shown in Table 6.
Comparison Between mapAlign and Minimap2 for Data Set 1 and Data Set 2 with GRCh38 As the Reference
From Table 6, we can see that mapAlign is faster than Minimap2 for both data set 1 and data set 2. Our methods use about 53% and 63% CPU time of Minimap2 for data set 1 and data set 2, respectively. Moreover, the alignment quality (in terms of both the identify and alignment scores) of our method is better than Minimap2 for data set 1 and data set 2. We have a slightly higher failure case than Minimap2. That is because these reads are not similar to the reference segments at all and they have very low alignment quality in terms of both the alignment score and identity score. We use a bigger memory than Minimap2 because we use a full reference k-mer index, and Minimap2 uses minimizer to build the reference k-mer index. We also use data sets from two different human individuals, data set 5 and data set 6, to do a comparison. The results are in Table 7. The alignment score and identity score for mapAlign and Minimap2 for data set 5 and data set 6 are given in Figures 2 and 3, respectively.
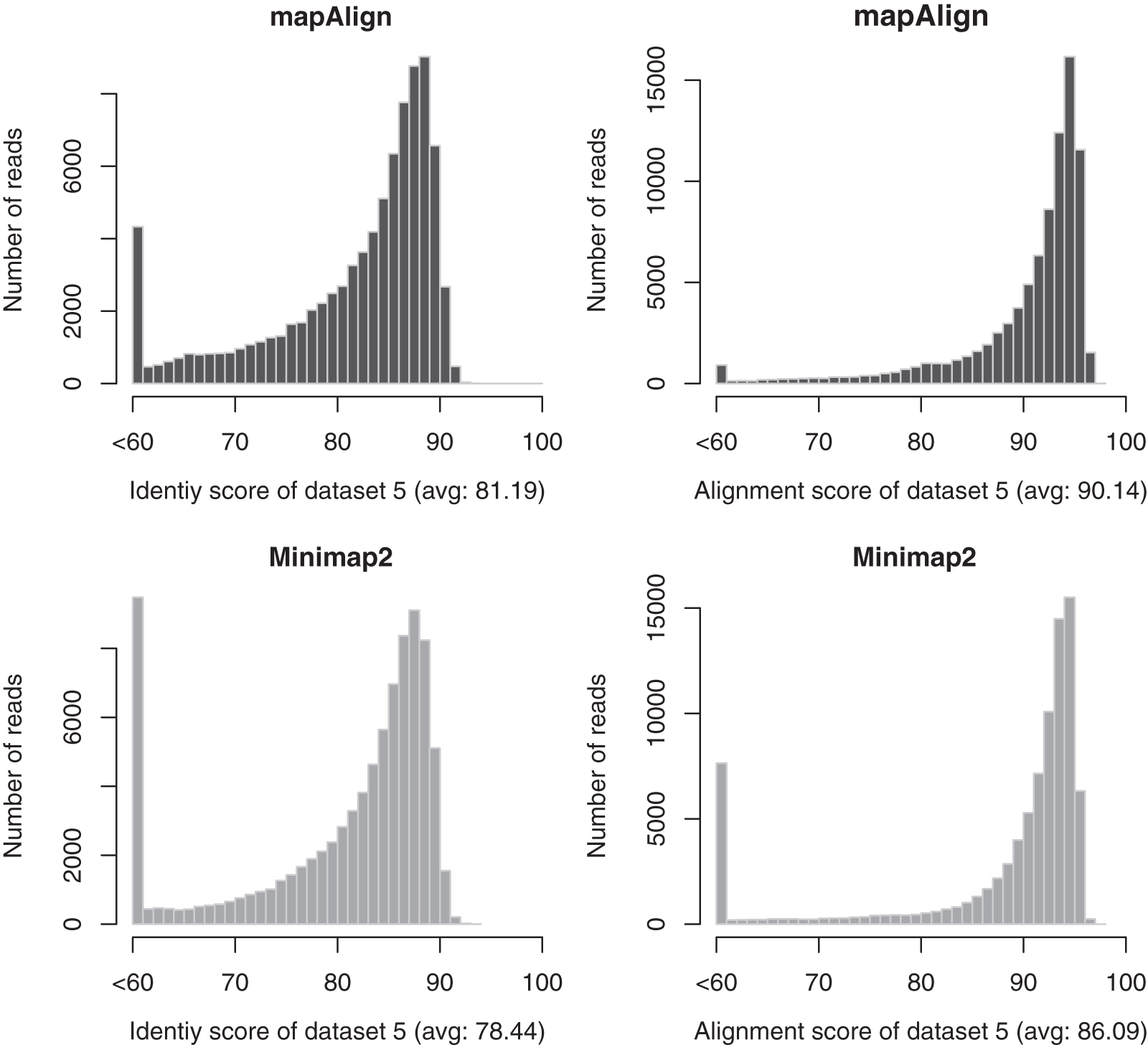
The comparisons of alignment score and identity score between mapAlign and Minimap2 for data set 5 with GRCh38 as the reference.
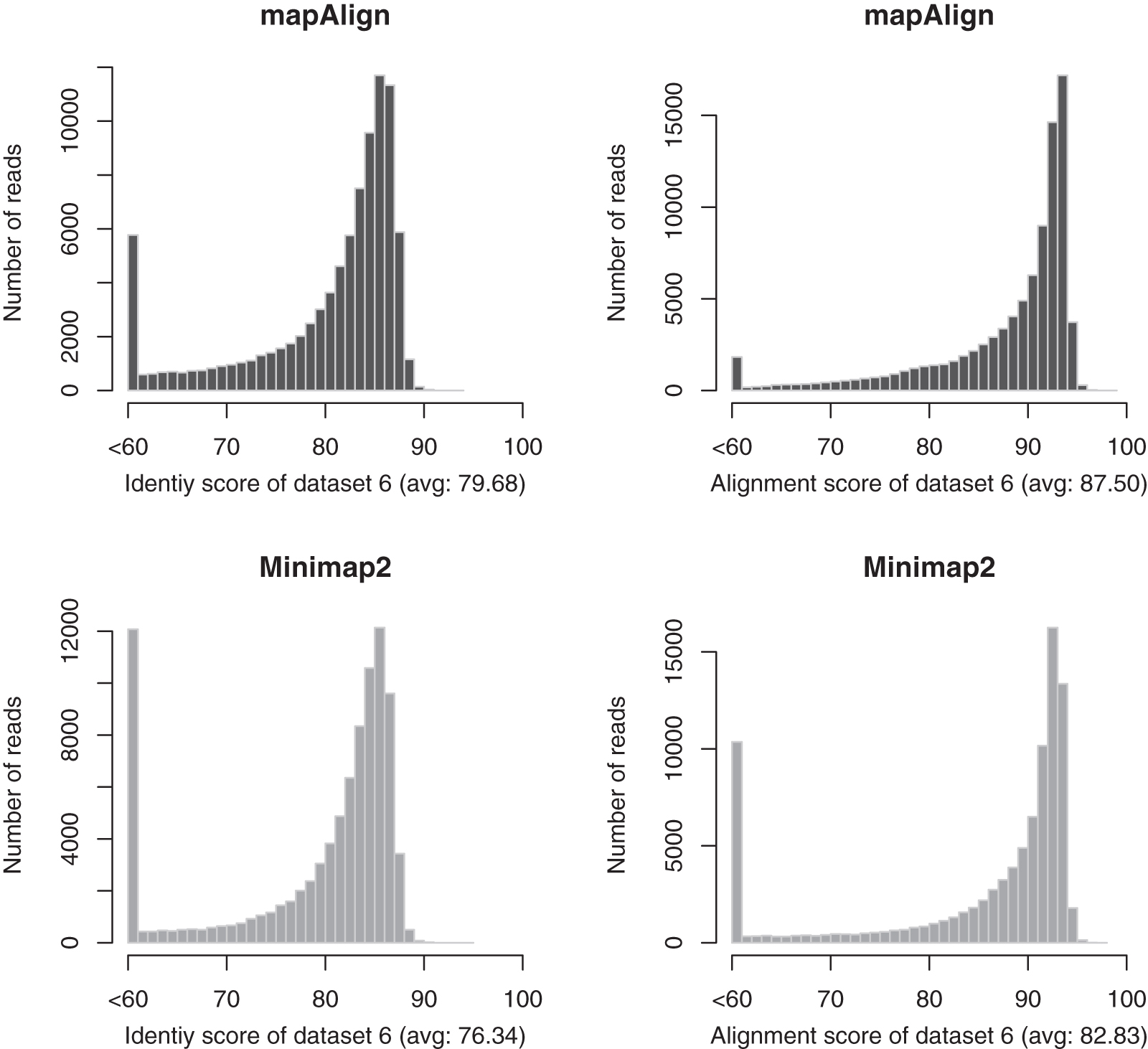
The comparisons of alignment score and identity score between mapAlign and Minimap2 for data set 6 with GRCh38 as the reference.
Comparison Between mapAlign and Minimap2 for Data Set 5 and Data Set 6 with GRCh38 As the Reference
4. Discussion
In this study, we present fast and accurate algorithms for mapping and aligning long reads and implemented them to form a software tool, mapAlign. For read mapping, we proposed the LCSDC problem and the approximate version, the
Our
There are many reasons that we can get better alignment quality. (1) After we get the matched pairs with a larger size k-mer (e.g., 16 or 15) from our
We believe our method still has room to further accelerate. For example, we could adopt the SIMD technique used in Minimap2. For the read mapping, we use a merge process to get the LCSDC, it involves a lot of sequential operation. Recently, the read mapping stage occupies a large proportion of the total running time. Thus, our future work will focus on the accelerating of read mapping. On the contrary, our merge process was similar to the sorted-set intersection in the databases’ area. Many works have been done for this area (Schlegel et al., 2011), proposing an efficient approach for sorted-set intersection by using the SIMD technique. This gives us many new clues for future work.
Footnotes
Author Disclosure Statement
The authors declare they have no competing financial interests.
Funding Information
This work is supported by a grant from the National Science Foundation of China (NSFC 61972329), and a GRF grant for the Hong Kong Special Administrative Region, China (CityU 11210119).