
Abstract
1. INTRODUCTION
Ribonucleic acid (RNA) is a biopolymer composed of nucleotides with bases adenine (A), cytosine (C), guanine (G), and uracil (U). The synthesis of an RNA molecule from its DNA template is initiated when the corresponding RNA polymerase binds to the DNA promoter region. RNA has been shown to serve diverse functions in a wide range of cellular processes, such as regulating gene expression and acting as an enzymatic catalyst (Storz, 2002; Collins and Penny, 2009), and has also recently been used as an emerging material for nanotechnology (Jasinski et al., 2017).
Computational prediction of RNA secondary structures given their sequences is often based on the estimation of changes in free energy, which postulates that thermodynamically an RNA strand will fold into a conformation that yields the minimum free energy (MFE) [see, e.g., Fallmann et al. (2017) for a review on the topic]. The energy of an RNA secondary structure can be modeled as the sum of energies of strand loops flanked by base pairs. The loop energy parameters have been measured experimentally and are detailed in a nearest neighbor parameter database (NNDB) (Turner and Mathews, 2009). Methods grounded in the thermodynamic framework, for example, the Zuker algorithm by Zuker and Stiegler (1981) and its extensions (Zuker, 1989; Mathews et al., 1999), can be used to compute pseudoknot-free MFE secondary structures effectively in a bottom-up manner. Recent attempts to extend the Zuker algorithm to find MFE secondary structures with certain classes of pseudoknots are also proposed (Rivas and Eddy, 1999; Akutsu, 2000; Dirks and Pierce, 2003; Reeder and Giegerich, 2004; Chen et al., 2009); however, finding MFE structures with pseudoknots given a general energy model is an NP-complete problem (Lyngsø and Pedersen, 2000).
The kinetic approach (Flamm et al., 2000) is an alternative way to study the RNA folding process. It models the folding as a random process where the additions/deletions of base pairs in the current structure are assigned probabilities proportional to the respective changes in free energy values. A folding pathway of a sequence is then generated by executing stochastic simulation (Mironov and Lebedev, 1993; Flamm et al., 2000; Dykeman, 2015). We refer to Marchetti et al. (2017) for a comprehensive review on stochastic simulation and recent work (Thanh et al., 2014, 2016; Marchetti et al., 2016; Thanh et al., 2017) for state-of-the-art stochastic simulation techniques. Each simulation runs on a given RNA sequence can produce a list of possible structures that it can fold into. Such dynamic view of RNA folding allows one to capture cases where local conformations are progressively folded to create metastable structures that kinetically trap the folding, thus complementing the prediction of equilibrium MFE structures produced by the thermodynamic approach.
The study of RNA structure formation often assumes that the folding process starts from a fully synthesized open strand, the denatured state. However, experimental evidence (Pan and Sosnick, 2006; Watters et al., 2016) has shown that RNA starts folding already concurrently with the transcription. The nucleotide transcription speed varies from 200 nt/s (nucleotides per second) in phages, to 20–80 nt/s in bacteria, and to 5–20 nt/s in humans (Pan and Sosnick, 2006). The RNA dynamics also occur over a wide range of time scales where base pairing takes about 10−3 ms; structure formation is about 10–100 ms; and kinetically trapped conformations can persist for minutes or hours (Al-Hashimi and Walter, 2008). One consequence of considering cotranscriptional folding is that the base pairs at the 5′ end of the RNA strand will form first, whereas the ones at the 3′ end can only be formed once the transcription is complete, which leads to structural asymmetries. Cotranscriptional folding can thus form transient structures that are only present for a specific time period and involved in distinct roles. For instance, gene expression when considering such transient conformations of RNA during cotranscriptional folding can exhibit oscillation behavior (Bratsun et al., 2005). We refer to the review by Lai et al. (2013) for further discussion on the importance of cotranscriptional effects.
In this work, we extend the kinetic approach to take into account cotranscriptional effects and pseudoknots on the folding of RNA secondary structures at single-nucleotide resolution. Our contribution is twofold. First, we explicitly consider the elongation of RNA during transcription as a primitive action in the model. The time when a new nucleotide is added to the current RNA chain is specified by the transcription speed of the RNA polymerase enzyme. The RNA strand in our modeling approach can elongate with newly synthesized nucleotides added to the sequence and fold simultaneously. To handle the transcription events, we propose an exact stochastic simulation method, the CoStochFold algorithm, to correct the folding pathway. Our method is thus able capture the effects of cotranscriptional folding at single-nucleotide resolution instead of approximating it as in previous approaches (Mironov and Kister, 1986; Flamm et al., 2000; Zhao et al., 2011; Proctor and Meyer, 2013; Hua et al., 2018). Second, our algorithm allows the formation of pseudoknots, which are important for understanding RNA functions. To cope with the challenge in evaluating the energy of pseudoknotted RNA structures, we adapt the NNDB model (Dirks and Pierce, 2003; Andronescu et al., 2010) to calculate their energy values. It is worth noting that determining a reasonable energy model for RNA structures with pseudoknots is still an open question (Lyngsø and Pedersen, 2000; Chen et al., 2009). However, the advantage of our strategy in comparison with other approaches, for example, adapting polymer theory in protein folding (Dill, 1999) to evaluate energy of pseudoknots (Isambert and Siggia, 2000), is that in the future when experimental data for pseudoknot parameters are established we can readily apply the simulation without revalidating parameters of the energy model. In addition, we facilitate the computation of energy of RNA structures with pseudoknots by employing a motif tree representation. This concept extends the coarse-grained tree representation of pseudoknot-free RNA structures, for example, Hofacker and Stadler (2005), to allow also pseudoknotted motifs.
The rest of the article is organized as follows. Section 2 reviews some background on kinetic folding of RNA. In Section 3, we present our work to extend the model of RNA folding to incorporate the transcription process and handle the formation of pseudoknots. Section 4 reports our numerical experiments on case studies. Concluding remarks are in Section 5.
2. BACKGROUND ON KINETIC FOLDING
Let Sn be a linear sequence of length n of four bases A, C, G, and U in which the 5′ end is at position 1 and the 3′ end is at position n. A base at position i may form a pair with a base at j, denoted by (i, j), if they form a Watson–Crick pair A-U, G-C, or a wobble pair G-U. A secondary structure formed by intramolecular interactions between bases in Sn is a list of base pairs
Let
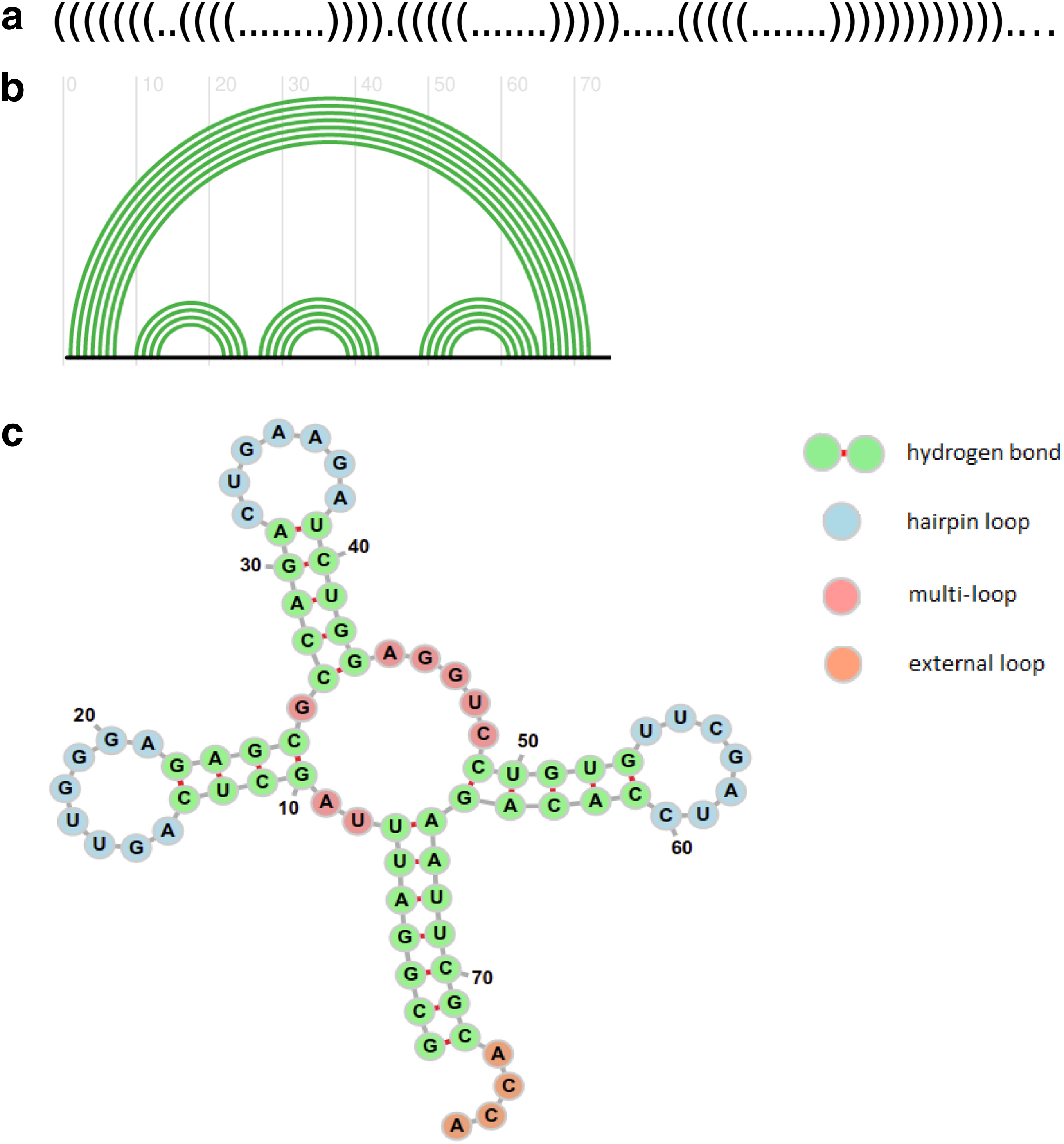
Representation of the tRNA molecule in
The free energy of x can be estimated by the nearest neighbor model (Mathews et al., 1999), in which the free energy of an RNA secondary structure is taken to be the sum of energies of components flanked by base pairs. Formally, for a base pair
A secondary structure x is thus uniquely decomposed into a collection of loops
where
Let
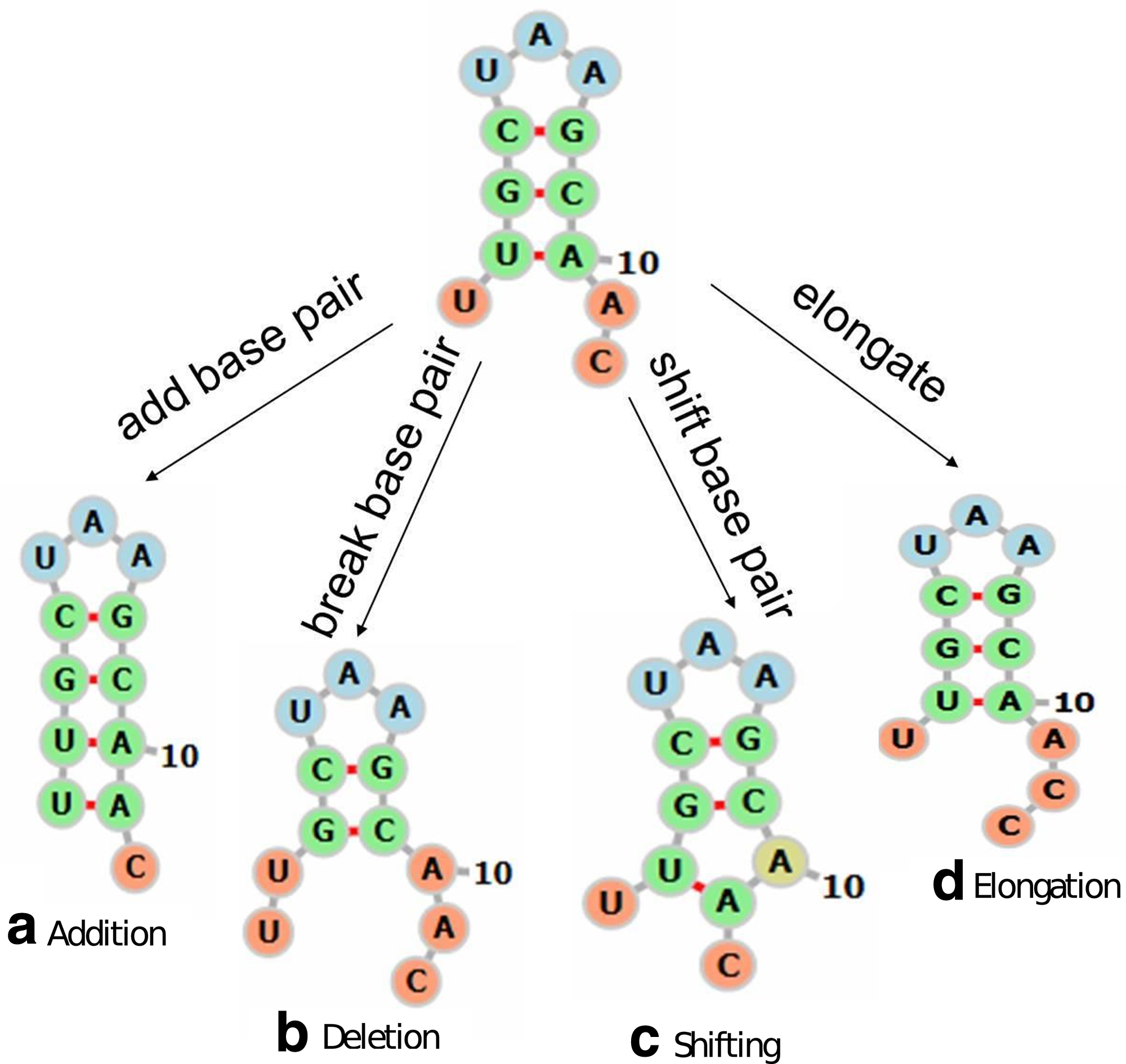
Extended move set consisting of
Addition: y is derived by adding a base pair that joins bases i and j in x that are currently unpaired and eligible to pair.
Deletion: y is derived by breaking a current base pair
Shifting: y is derived by shifting a base pair
Let
where T is absolute temperature in Kelvin (K),
Let
Analytically solving Equation (3) requires to enumerate all possible states x and their neighbors y. The size of the state space
Let
where
3. COTRANSCRIPTIONAL KINETIC FOLDING OF RNA
The folding of an RNA strand adapts immediately to new nucleotides synthesized during the transcription. The kinetic approach described in Section 2 cannot capture the effects of such cotranscriptional folding because it considers only interactions between bases already present in the sequence. We outline in this section an approach to incorporating these effects in the simulation. The transcription process is explicitly taken into account by extending the move set with the new operation of elongation. Our extended move set thus comprises four operations: addition, deletion, shifting, and elongation. The first three operations are defined as in the previous section. In elongation, the current RNA chain increases in length and a newly synthesized nucleotide is added to its 3′ end. Figure 2 illustrates the extended move set.
Under the extended move set, we define two event types: folding event and transcription event. A folding event is an internal event that occurs when one of the three operations, addition, deletion, or shifting, is applied to a base pair of the current sequence. A transcription event happens when the elongation operation is applied. It is an external event whose rate is specified by the transcription speed of the RNA polymerase enzyme. The occurrences of transcription events break the Markovian property of transitions between conformations. This is because when a new nucleotide is added to the current RNA conformation, the number of next possible conformations increases. The waiting time of the next folding event also changes, and thus, a new folding event has to be recomputed.
Algorithm 2 outlines how the CoStochFold algorithm handles this situation. The key element of CoStochFold (lines 8–15) is a race where the event having the smallest waiting time will be selected to update the current RNA conformation. More specifically, suppose the current structure is x at time t. Let
Thus, given current time t, the next folding event will occur at time
We remark that one can easily extend Algorithm 2 to allow modeling
3.1. Handling pseudoknots
This section extends the CoStochFold algorithm to include structures with pseudoknots during the enumeration of neighbor structures (see step 5, Algorithm 2). A pseudoknot occurs if there exists a crossing between two base pairs. Here, we restrict to the two most common pseudoknots: the H-type and K-type (kissing hairpin) (Reidys et al., 2011). We use the extended dot-bracket notation, that is, augment the original dot bracket with additional types of bracket pairs, for example, [],

Let
where
To facilitate the evaluation of the energy of an RNA structure x with pseudoknots, we first parse x to closed regions (Rastegari and Condon, 2007). A set of bases

An example of a motif tree.
4. NUMERICAL EXPERIMENTS
We illustrate the application of our cotranscriptional kinetic folding method on four case studies: (1) the Escherichia coli signal recognition particle (SRP) RNA (Watters et al., 2016), (2) the switching molecule (Flamm et al., 2000), (3) the Beet soil-borne virus (Taufer et al., 2008), and (4) the SV-11 variant in Q
4.1. Signal recognition particle RNA
This section studies the process of structural formation of the E. coli SRP RNA during transcription. SRP is a 117 nt long molecule, which recognizes the signal peptide and binds to the ribosome locking the protein synthesis. Its active structure is a long helical structure containing interspersed inner loops (see S3 in Fig. 5). Experimental work (Watters et al., 2016) using SHAPE-seq techniques has suggested a series of structural rearrangements during transcription that ultimately result in the SRP helical structure. In particular, the 5′ end of SRP forms a hairpin structure during early transcription. The structure persists until the transcript reaches a length of 117 nt. The unstable hairpin then rearranges to its active structure. Figure 5 depicts three structural motifs at 25 nt (S1), 86 nt (S2), and 117 nt (S3), respectively, in the formation of SRP. Specifically, the hairpin motif S1 emerges at transcript length 25 nt, and the transcript then continues elongating to form structure S2 at length 86 nt. When reaching transcript length 117 nt, SRP rearranges into its persistent helical conformation S3.

The folding pathway of secondary structures of the Escherichia coli SRP RNA. The hairpin motif S1
We validated the prediction of the CoStochFold algorithm against the experimental work in Watters et al. (2016). To do that, we performed 10,000 simulation runs of the algorithm to fold SRP cotranscriptionally. The average transcription speed was set to 5 nt/s. Figure 6 shows the frequency of occurrences of the considered structures during the simulated time of 30 seconds. Kinetic folding starting from the denatured state was carried out by the StochFold algorithm, whereas cotranscriptional folding was conducted by the CoStochFold algorithm. The plot on the left shows the cotranscriptional folding of SRP and the plot on the right presents the folding of SRP starting from the denatured state. The figures clearly show that the CoStochFold algorithm can capture the folding pathway of SRP. Specifically, the hairpin motif S1 starts to form at about t = 4 s when the transcript length is 20 nt and peaks at about t = 8 s when 40 nt have been transcribed. At about t = 18 s, Structure S2 appears and then rearranges to S3 at about t = 24 s. We note that in the simulated folding without considering transcription only the conformation S3 is encountered.

Prediction of the structural formation of SRP. Left: cotranscriptional folding. Right: folding from denatured state without transcription. The frequency of occurrence of a motif on the y-axis is computed as the numbers of occurrences over total 1000 simulation runs. Time on the x-axis is in seconds of simulated time.
4.2. Switching molecule
We consider the dynamic folding of an artificial RNA sequence S = “GGCCCCUUUGGGGGCCAGACCCCUAAAGGGGUC” (Flamm et al., 2000). Two stable conformations of the sequence are: the MFE structure x = “((((((((((((((.….))))))))))))))” (−26.20 kcal) and a suboptimal structure y = “((((((.…)))))).((((((.…))))))” (−25.30 kcal). We use this example to demonstrate how by tuning the transcription speed we can change the ratio of occurrences of structures x and y. Here, we focus on the number of first-hitting time occurrences of a target structure. The number of first-hitting time occurrences of a structure in a time interval divided by the total number of simulation runs approximates the first-passage time probability of the structure, that is, its folding time (Flamm et al., 2000).
Figure 7 plots the number of first-hitting time occurrences of the MFE structure x and the suboptimal y with varying transcription speeds. We performed 10,000 simulation runs of the CoStochFold algorithm on the sequence S in which each simulation ran until a target structure was observed or the ending time
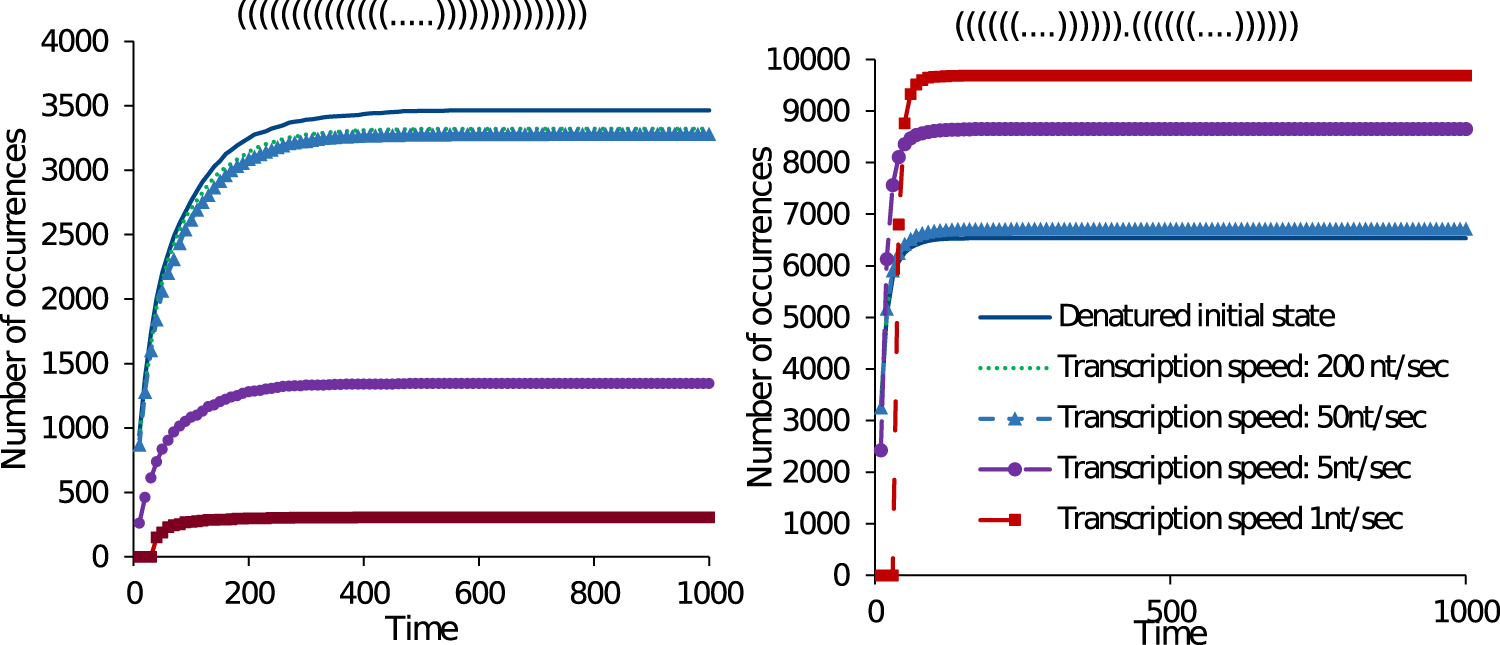
Cumulative first-hitting time occurrences of MFE structure x = “((((((((((((((.….))))))))))))))” (−26.20 kcal, left) and suboptimal y = “((((((.…)))))).((((((.…))))))” (−25.30 kcal, right). Time on the x-axis is in seconds of simulated time. MFE, minimum free energy.
Figure 8 compares the total number of first-hitting time occurrences of the MFE structure x with respect to the suboptimal conformation y up to time

Total number of occurrences of MFE structure x = “((((((((((((((.….))))))))))))))” (−26.20 kcal) and suboptimal y = “((((((.…)))))).((((((.…))))))” (−25.30 kcal) with simulated time
4.3. Beet soil-borne virus
We use the beet soil-borne virus S = “CGGUAGCGCGAACCGUUAUCGCGCA” from the PseudoBase++ database (Taufer et al., 2008) to demonstrate the application of our simulation in predicting RNA structures with pseudoknots. The folding of the sequence S was simulated with 10,000 runs. We evaluated the energy of pseudoknots using the energy parameters from Andronescu et al. (2010), which were estimated by fitting the standard NNDB parameters by Mathews et al. (1999) and pseudoknotted parameters by Dirks and Pierce (2003) over a large data set of both pseudoknotted and pseudoknot-free secondary structures. We compare two simulation settings: (1) cotranscriptional folding of S with transcription speed 200 nt/s and (2) the folding starting from the denatured initial state (i.e., a fully synthesized open strand). Figure 9 depicts the occurrence frequency of the H-type pseudoknotted structure C1 = “.(((.[[[[[[)))…]]]]]].” with an energy of

Structural formation of the Beet soil-borne virus in
Figure 9a and 9b clearly show that the dominant structure of the beet soil-borne virus sequence S is the H-type pseudoknotted structure C1. We also observe from these figures that the folding starting from the denatured state misses the formation of intermediate structures C2 and C3, which appear in the cotranscriptional folding. After the transcription phase, intermediate structures will rearrange to C1 and remain in this stable form. Figure 9a shows that the frequency of C1 is >82% in the simulation.
We conclude this section with a note about the energy parameters for RNA structures with pseudoknots. In particular, we also simulated the beet soil-borne virus S with the energy model by Reidys et al. (2011), which is another extension of the NNDB model for pseudoknots. The occurrence frequency of pseudoknotted structure C1 estimated by the Reidys et al. (2011) model was significantly lower than that by the Andronescu et al. (2010) model. This prediction discrepancy is because the energy model by Reidys et al. (2011) penalizes the formation of pseudoknots significantly more than the model by Andronescu et al. (2010). In fact, all pseudoknotted structures will be unfavorable with such high penalties for the pseudoknots. An interesting prediction from our cotranscriptional folding simulation using both energy models is the occurrence of the intermediate hairpin structure C3. The persistence of C3 before rearranging to the pseudoknot C1 depends on how much penalty is applied to the formation of pseudoknots.
4.4. SV-11
SV-11 is a 115 nt long RNA sequence. It is a recombinant between the plus and minus strands of the natural Qβ template MNV-11 RNA (Biebricher and Luce, 1992). The result of the recombination is a highly palindromic sequence whose most stable secondary structure is a long hairpin-like structure, the MFE structure in Figure 10a. The MFE structure, however, disables Qβ replicase because its primer regions are blocked. Experimental work (Biebricher and Luce, 1992) has shown that an active structure of SV-11 for replication is when it folds into a metastable conformation depicted in Figure 10b. This is a hairpin–hairpin–multi-loop motif with open primer regions that serve as templates for replication. Transition from the metastable structure to the MFE structure has been observed experimentally but is rather slow (Biebricher and Luce, 1992), indicating long relaxation time to equilibrium.
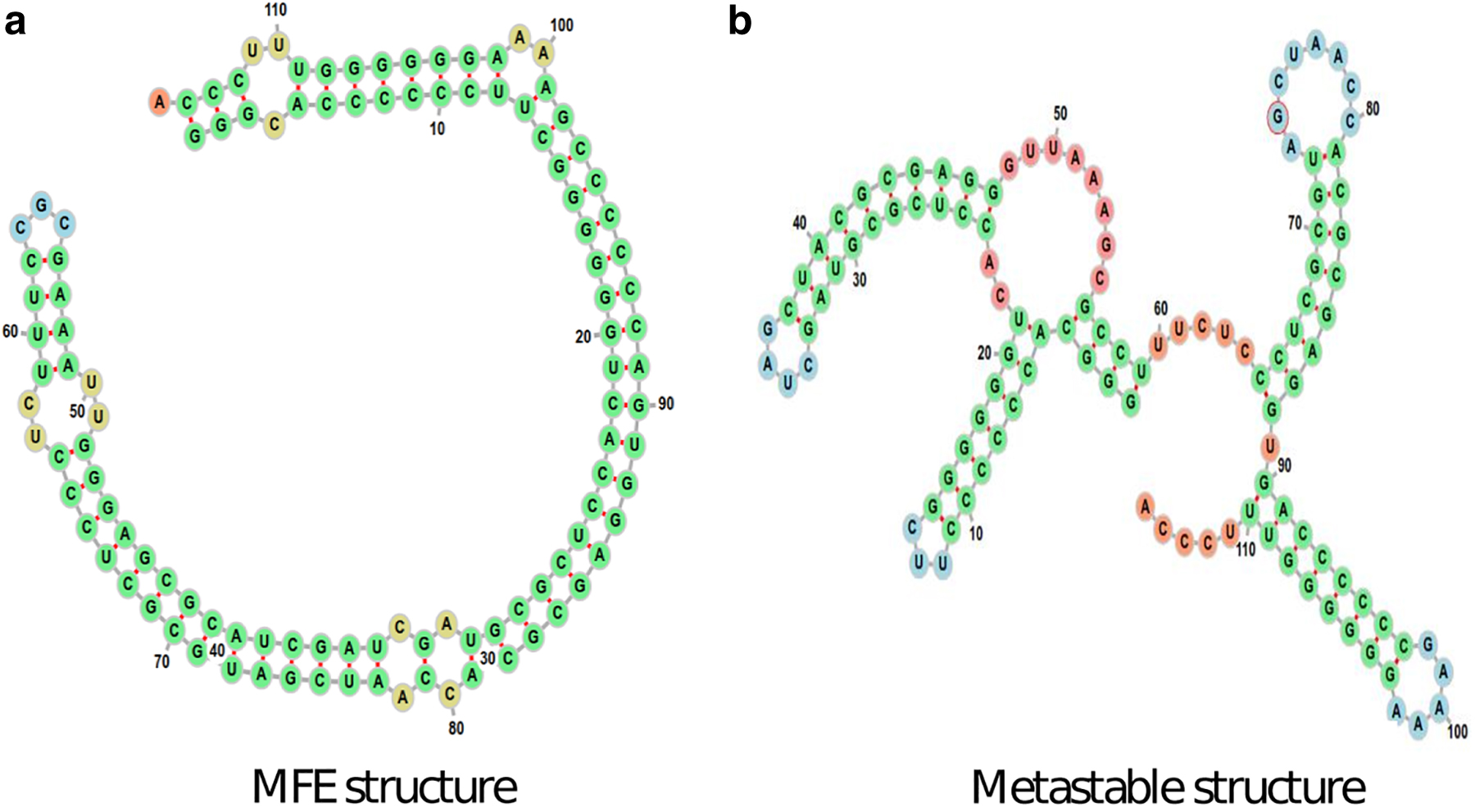
SV-11 with two conformations:
We plot in Figure 11 the energy versus occurrence frequency of structures by the cotranscriptional folding of SV-11. The result is obtained by 10,000 simulation runs of our CoStochFold algorithm for t = 50 simulated seconds and average transcription speed 5 nt/s. To determine the frequency of occurrence of a structure, we discretize the simulation time into intervals and record how much time was spent in each structure within each interval. The frequency of occurrence of a structure in each time interval is then averaged over 10,000 runs. The figure shows that the folding favors metastable structures and disfavors the MFE structure. In particular, cotranscriptional folding quickly folds SV-11 to its metastable conformations with the mode of the energy distribution at about −63 kcal.
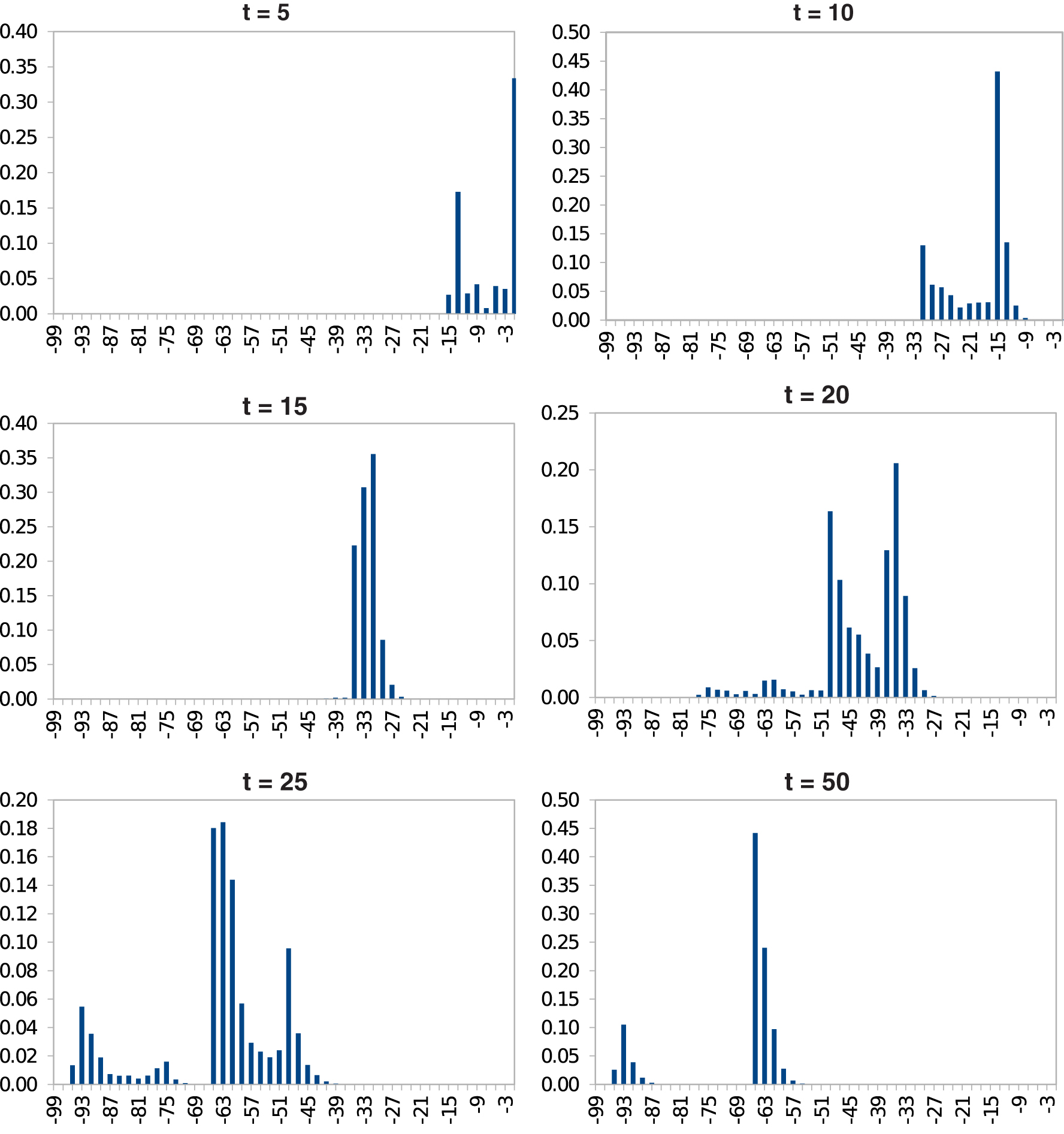
Cotranscriptional folding of SV-11. The x-axis denotes the energy level in kcal, and the y-axis shows the frequency of structures at a given energy level.
Figure 12 shows the long-term occurrence frequencies of structures at different energy levels in the SV-11-folding, and Figure 13 compares the occurrence frequencies of the specific metastable structure depicted in Figure 10b, with the MFE structure and two randomly selected suboptimal structures in the energy level of MFE structure. Figure 13 shows that the SV-11 molecule interestingly prefers the metastable structure over the MFE structure. Specifically, the metastable structure in the cotranscriptional folding regimen is in the time interval [0, 10,000] about 10-fold more frequent than the MFE structure.

Frequency of structures in folding SV-11.
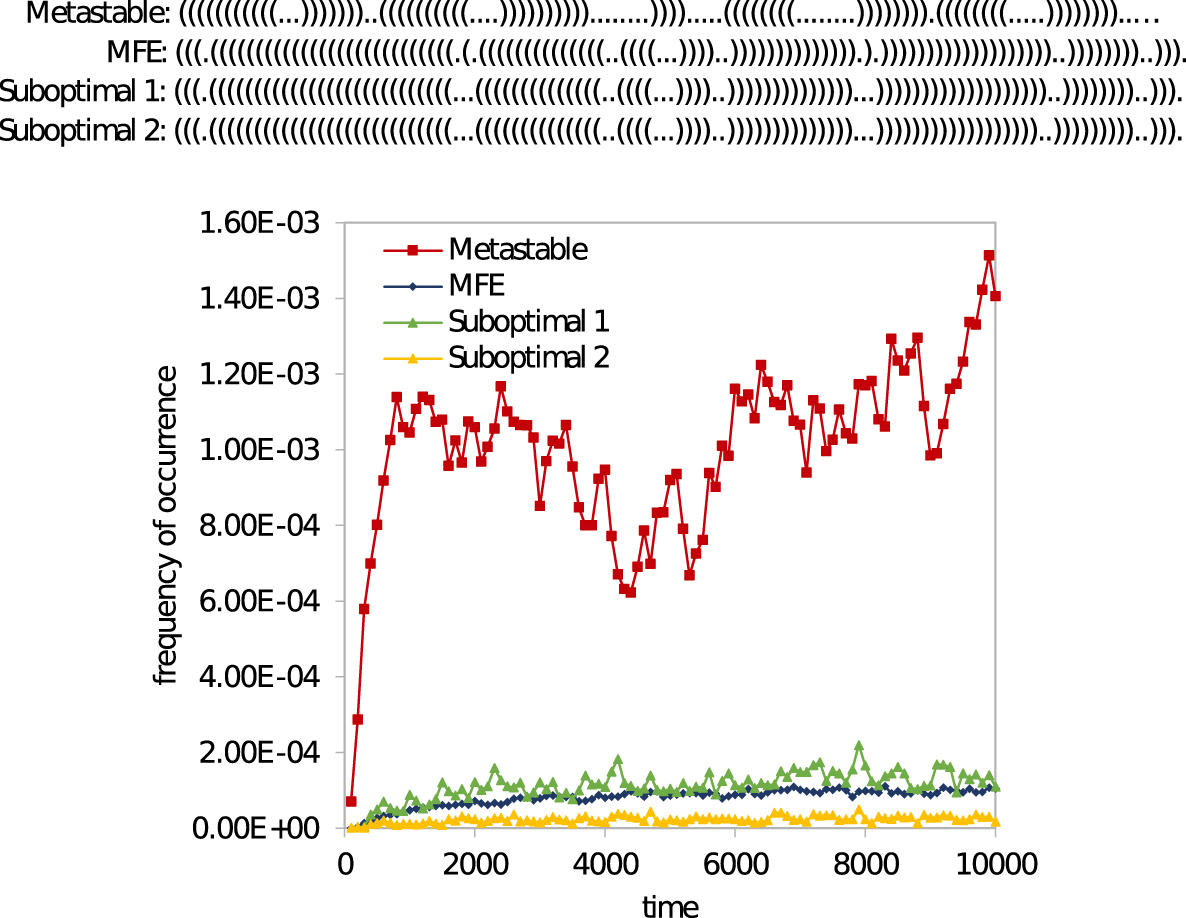
Frequency of the metastable structure in comparison with the MFE structure and two randomly selected suboptimal structures in the locality of the energy level of MFE.
4.5. Simulation performance
This section reports the performance of our stochastic folding algorithm with RNA sequences of varying lengths from 25 to 5000 nt. To estimate the computational cost of a single simulation move, we executed 10 independent simulation runs of 1000 simulation steps, each with a random sequence of the given length. The average runtime for each sequence length was computed and then divided by the number of simulation steps to assess the single-step computation cost.
Figure 14 plots the resulting estimated single-step computational cost of our folding algorithm in two settings: (1) simulation without pseudoknots, executed on Intel an i5-7300U dual-core CPU with a clock speed of 2.6 GHz, on the left and (2) simulation with pseudoknots, executed on an Intel i5-8365U quad-core CPU with a clock speed of 1.6 GHz, on the right. As witnessed by the figure, the simulation is quite computation intensive, especially for long sequences. For example, the simulation without pseudoknots for a sequence of length 1000 nt took on average 0.1 s of processor time per simulation step. Thus, a single simulation run of 1000 simulation steps would take on average 100 s, and 10,000 repeats of this would take 106 s, that is, 11.6 days of processor time.
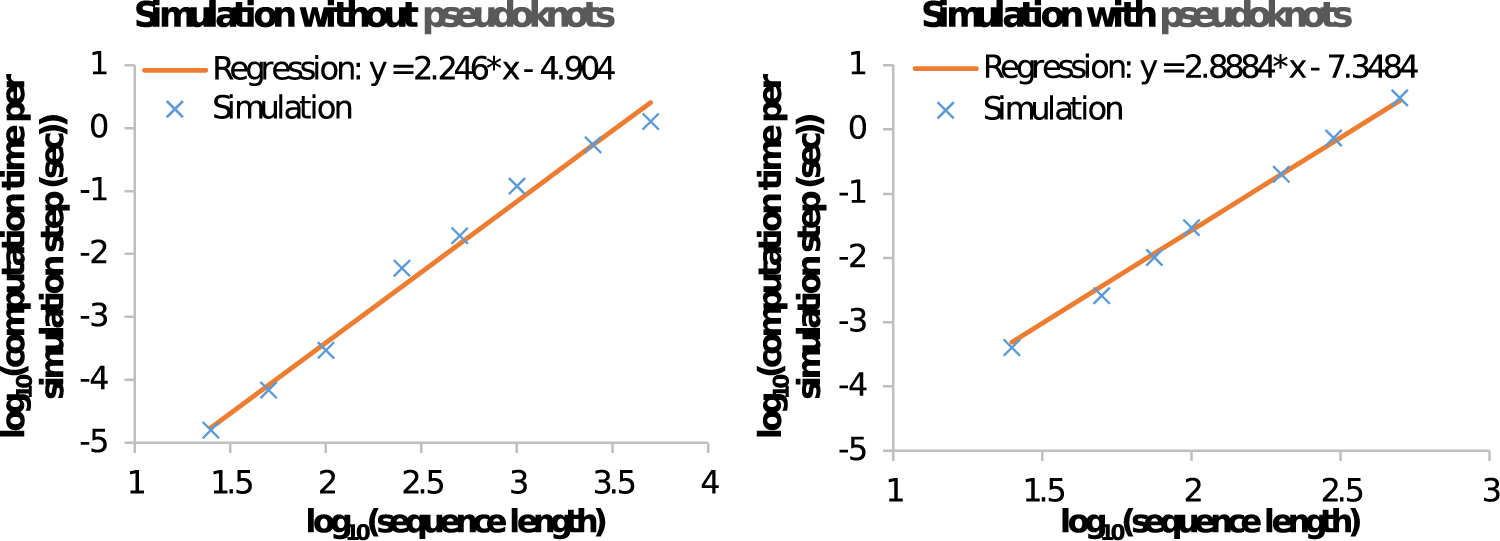
Computational runtimes of stochastic folding with sequences of varying lengths. Left: simulation without pseudoknots. Right: simulation with pseudoknots. Values on the x-axis and y-axis are in logarithmic scale.
The single-step computational cost increases with increasing sequence length: the runtime in the case of a pseudoknot-free simulation for sequences of length 5000 nt is about 11 times higher than for sequences of length 1000 nt. This increase is mostly due to the quadratically increasing number of possible moves in the locality of a conformation. Our detailed breakdown analysis of the computational cost of simulations shows that the cost of enumerating the possible moves contributes >95% of the total cost in each simulation step. (Note that the cost of enumerating the moves depends on both the number of possible moves and the algorithmics of the enumeration process.) The regression lines depicted in Figure 14 indicate that the computational cost per single move of our folding algorithm without pseudoknots grows as
5. CONCLUSIONS
We propose a kinetic model of RNA folding that takes into account the elongation of an RNA chain during transcription as a primitive structure-forming operation alongside the common base pairing operations. We developed a new stochastic simulation algorithm CoStochFold to explore RNA structure formation, including pseudoknots, in the cotranscriptional folding regimen. We showed through numerical case studies that our method can quantitatively predict the formation of (metastable) conformations in an RNA folding pathway. The simulation method thus promises to offer useful insights into RNA folding kinetics in real biological systems. However, it also poses a great computational challenge for long sequences due to the huge number of possible moves in the locality of a conformation. Furthermore, many simulation runs must be performed to obtain a reasonable statistical estimation of the system dynamics. Several improvements are possible in future work. For instance, we can reduce the enumeration of possible moves by localizing the computation. The motif tree, a coarse-grained representation for pseudoknotted structures developed in the article, could be useful also in this context. We decompose an RNA structure into motifs and then enumerate new conformations related to each motif. To reduce the cost for executing many simulation runs, we can employ high-performance computing to run simulations in parallel.
Footnotes
AUTHOR DISCLOSURE STATEMENT
No competing financial interests exist.
FUNDING INFORMATION
This work has been supported by Academy of Finland grant no. 311639, “Algorithmic Designs for Biomolecular Nanostructures (ALBION).” The work of V.H.T. has been partially done when he was at Aalto University.