
Abstract
The detrimental effect of coronavirus disease 2019 (COVID-19) pandemic has manifested itself as a global crisis. Currently, no specific treatment options are available for COVID-19, so therapeutic interventions to tackle the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection must be urgently established. Therefore, cohesive and multidimensional efforts are required to identify new therapies or investigate the efficacy of small molecules and existing drugs against SARS-CoV-2. Since the RNA-dependent RNA Polymerase (RdRP) of SARS-CoV-2 is a promising therapeutic target, this study addresses the identification of antiviral molecules that can specifically target SARS-CoV-2 RdRP. The computational approach of drug development was used to screen the antiviral molecules from two antiviral libraries (Life Chemicals [LC] and ASINEX) against RdRP. Here, we report six antiviral molecules (F3407-4105, F6523-2250, F6559-0746 from LC and BDG 33693278, BDG 33693315, LAS 34156196 from ASINEX), which show substantial interactions with key amino acid residues of the active site of SARS-CoV-2 RdRP and exhibit higher binding affinity (>7.5 kcalmol−1) than Galidesivir, an Food and Drug Administration-approved inhibitor of the same. Further, molecular dynamics simulation and Molecular Mechanics Poisson-Boltzmann Surface Area results confirmed that identified molecules with RdRP formed higher stable RdRP-inhibitor(s) complex than RdRP-Galidesvir complex. Our findings suggest that these molecules could be potential inhibitors of SARS-CoV-2 RdRP. However, further in vitro and preclinical experiments would be required to validate these potential inhibitors of SARS-CoV-2 protein.
1. Introduction
Recently, a novel strain of coronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been identified as the cause for coronavirus disease 2019 (COVID-19). The advent of this new extremely infectious and virulent strain of coronavirus (SARS-CoV-2) has posed one of the greatest challenges for the scientific community around the world. Coronaviruses belong to the Coronoviridae family and are further divided into four genera, that is, alpha, beta, gamma, and delta coronavirus (Li, 2016). The alpha and beta members are responsible for mammalian infection, whereas gamma and delta members are responsible for infection in birds. Alpha coronaviruses, such as 229E (HCoV-229E) and NL63 (HCoV-NL63), and those belonging to the beta-coronavirus genus, such as OC43 (HCoV-OC43) and HKU1 (HCoV-HKU1), are established human pathogens with global prevalence (Tang et al., 2015; Wu et al., 2020a).
SARS-CoV-2 belongs to the beta-coronavirus genus, which is closely similar to SARS-CoV and middle east respiratory syndrome (MERS)-CoV (Ibrahim et al., 2020). Genome organization of all coronaviruses is similar and has 5′ and 3′ untranslated regions responsible for the coding of genes for spike, ORF1ab, membrane, enclosure, and nucleocapsid protein (Lai et al., 1985). Two-third of the coronavirus genome is occupied by the polycistronic ORF1ab sequence encoding a polyprotein “replicase” from the ORF1a and ORF1b genes (Woo et al., 2005, 2007, 2010). ORF1ab encodes a 3C-like protease (3CLpro) and a Papain-like protease (PLpro), which cleave the polyprotein replicase, at consensus cleavage sites, into 15–16 non-structural proteins (nsps). Some of these are essential ones, namely PLpro (nsp3), 3CLpro (nsp5), and RdRP (nsp12), which play indispensable roles in viral replication. nsp13 is a helicase responsible for nucleoside triphosphate-dependent (NTP-) unwinding of duplex oligonucleotide (Woo et al., 2005, 2010; Jia et al., 2019). Barring retroviruses, the majority of RNA viruses require RdRP for the replication of corresponding viral genomes and their subsequent transcription, thereby making this enzyme a mainstay for the survival and propagation of these viruses (Lai, 2005).
As mentioned earlier, RdRP is a product of polyprotein cleavage and consists of 1a and 1ab translated, respectively, from ORF1a and ORF1ab (Kirchdoerfer and Ward, 2019). The RdRP has a catalytic core strongly resembling a human-right hand with distinct palm, thumb, and finger domains (Fig. 1) (Wu et al., 2015; Smertina et al., 2019). The core protein is a single chain of about 900 amino acids with limited activity. Association with other key subunits (nsp7/nsp8 in a pre-complex and an additional nsp8) enhances this activity and constitutes the minimal complex sufficient for the effective functioning of SARS-CoV-2 (Ahn et al., 2012; Subissi et al., 2014; Kirchdoerfer and Ward, 2019). The active site of SARS-CoV-2 RdRP, which plays a central role in viral replication and transcription, is conserved among the RNA viruses (Wang et al., 2020a). Thus, targeting the active site of RdRP could be a potential antiviral strategy (Hansen et al., 1997; Ago et al., 1999; Lesburg et al., 1999; Xu et al., 2003; Kinsella et al., 2004; Gerlach et al., 2015; Liang et al., 2015). Owing to their anti-RdRP activities, many drugs such as Galidesivir, Sofosbuvir, Remdesivir, Ribavirin, etc. are being clinically tested against different kinds of viruses, such as Zika, Hepatitis C, SARS-CoV-2, and Dengue (Markland et al., 2000; McQuaid et al., 2015; Elfik, 2016; Elfiky and Ismail, 2019; Elfiky et al., 2017; Ezat et al., 2019; Wang et al., 2020a; Kumar et al., 2021).

Schematic representation of the SARS-CoV-2 genome. The 30 kbp long genomic RNA of SARS-CoV-2 virus can be broadly divided into two parts: 20 kbp 5′ section encoding for nsps, whereas the remaining 10 kbp long 3′ stretch getting translated into structural and accessory proteins. The nsps come out of two open reading frames, ORF1a and ORF1b. Two polypeptides, pp1a and pp1ab, come out of these two ORFs. A frameshift at the junction of ORF1a and ORF1b leads to the translation of 790 kDa polypeptide pp1ab, which gets processed into nsps1–16, without nsp11. nsp11 is one of the products from the processing of 490 kDa pp1a, with the rest being nsps1–10. nsps1–3 arise due to proteolytic cleavage of either pp1a or pp1ab by PL proteinase, whereas the rest of the nsps, except nsp12, result from the cleavages catalyzed by 3CL proteinase. nsp12 is the functional Rdrp and results from an N- and C-terminal cleavage by 3CL and PL proteinases, respectively. nsp12 consists of N-terminal NiRAN and C-terminal RdRP domains. The NiRAN domain is interspersed with a β-hairpin motif closer to its N-terminus. The Rdrp domain consists of fingers, palm, and thumb motifs in the N to C direction. Surface (S), envelope (E), membrane (M), and nucleocapsid (N) proteins are the structural and accessory building blocks arising from this viral genome. 3CL, 3C-like protease; nsps, non-structural proteins; ORF, open reading frame; PL, papain-like protease; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
In the context of SARS-CoV-2, several antiviral drugs have been clinically tested for repurposing, with the obvious reason to cut down on the de novo drug finding time and cost, but with limited success (Beigel et al., 2020; Boulware et al., 2020; Geleris et al., 2020; Pan et al., 2020). Contrastingly, some reports also indicate the efficacy of various antiviral drugs, purified natural compounds, and herbal medicines against different SARS-CoV-2 proteins (Elfiky, 2020a; Anand et al., 2021). Nevertheless, the repurposing of known drugs remains the best choice for identifying a cure to a novel disease under time constraint, as in case of the current COVID-19 pandemic (Singh et al., 2020). Nowadays, computational approaches, such as screening and molecular dynamics (MD), are commonly employed in the early stages of drug discovery to identify potential therapeutic compounds (Kumar et al., 2021). Therefore, this study was designed for exhaustive mining of the two prominent libraries specific for antiviral molecules.
Employing the computational approach, the current study focuses on the identification of potential antiviral compounds that can inhibit SARS-CoV-2 RdRP, the enzyme required for the overall survival of the virus. We have used the Food and Drug Administration (FDA) approved drugs as positive controls for the identification of novel molecules potent in inhibiting SARS-CoV-2 RdRP. The antiviral molecules of Life chemicals (LC) and ASINEX libraries were screened against the RdRP. Totally six antiviral molecules (molecules with IDs F3407-4105, F6523-2250, F6559-0746 from LC antiviral library, and those with IDs BDG 33693278, BDG 33693315, LAS 34156196 from ASINEX antiviral library) were selected, which were found to have higher binding affinities than FDA-approved SARS-CoV-2 RdRP inhibitors, forming a better network of molecular interactions at the active site of RdRP. Our findings provide a reliable base for the development of new drugs that can inhibit SARS-CoV-2 RdRP, ultimately implicating a reduction in COVID-19 infection.
2. Methods
2.1. Retrieval of three-dimensional structure of RdRP and multiple sequence alignment
The crystal structure of nsp12 (RNA-dependent RNA polymerase [RdRP]) protein of SARS-CoV-2 (PDB ID: 7C2K) was retrieved from the RCSB-PDB database (Wang et al., 2020b) and used in this study. Further, the conserved motifs and active site of RdRP were assessed by generating the multiple sequence alignment (MSA) from SARS-CoV-2 with its homologs from other human coronaviruses (HCoV) using Clustal omega and illustrated using ESPript3 (Gouet et al., 1999; Sievers et al., 2011).
2.2. Molecular docking of existing drug against RdRP of SARS-CoV-2
Nucleotide analog drugs, such as Galidesivir, Peniciclovir, Favipiravir, Remdesivir, Ribavirin, and Sofosbuvir, are reported to efficiently inhibit replication of SARS-CoV-2 in cell-based assays (Elfiky, 2020a; Yin et al., 2020). Therefore, these drugs were selected as reference molecules to find novel antiviral compounds. The structures of these drugs (PDB ID: UA2, GE6, PE2, F86, 6CG, and RBV, respectively, for Galidesivir, Favipiravir, Peniciclovir, Remdesivir, Sofosbuvir, and Ribavirin) were downloaded from the RCSB PDB database and processed by adding polar hydrogens and gasteiger charges.
The protein structure of RdRP of SARS-CoV-2 (PDB ID: 7C2K) was prepared by adding the essential hydrogen atoms and Kollman charges using the AutoDock Tools. The molecular docking was performed by using AutoDock Tools and AutoDock Vina (Morris et al., 2009; Trott and Olson, 2010; Saraswat et al., 2021). The grid box for molecular docking covered the active site residues (Arg553, Arg555, Asp618, Asn691, Ser759, Asp760, and Asp761) of the protein. The grid box dimensions and center point coordinates were set as 39 × 54 × 31 Å and 135.6, 138.6, and 140.5, respectively, with a default spacing value of 0.375 Å. Lamarckian Genetic Algorithm (GA) was used, and the number of GA runs were raised from 10 to 100. Based on the binding energy score and interactions with the residues of catalytic domain of SARS-CoV-2 RdRP, the drug molecule with the maximum inhibitory potential was identified.
2.3. Virtual screening of antiviral compounds
The virtual screening was performed by using the AutoDock Vina in PyRx0.8 platform (Trott and Olson, 2010; Dallakyan and Olson, 2015). Two antiviral libraries (LC [https://lifechemicals.com/] and ASINEX database [www.asinex.com/]) were used to find antiviral molecules against RdRP, which would be more potent than the FDA-approved drugs. The antiviral molecules of these libraries are the structural analogs of the nucleoside-based FDA-approved antiviral drug molecule such as favipiravir, sofosbuvir, lopinavir, ribavirin, galidesivir, and remdesivir. The antiviral libraries were retrieved in SDF format. Ligands were energy minimized by Universal Force Field and converted to .pdbqt format by using Open Babel in Pyrx 0.8 for virtual screening (O'Boyle et al., 2011). The grid map was centered on the active site residues (Arg553, Arg555, Asp618, Asn691, Ser759, Asp760, and Asp761) with the dimension of 39 × 54 × 31 Å. Each molecule from both libraries was docked into the active site of RdRP and scored for binding affinities in kcal/mol. The top-ranked molecules with higher binding affinities than FDA-approved drugs were evaluated by using PyMOL 2.1.1 (DeLano, 2002).
2.4. Re-docking using GA of top hits
The molecular docking of six lead compounds (F3407-4105, F6523-2250, F6559-0746, BDG 33693278, BDG 33693315, and LAS 34156196) obtained from the virtual screening was docked with RdRP protein by using AutoDock Tools (Morris et al., 2009). The essential hydrogen atoms and Kollman charges were added to the target protein structure by using Molecular Graphics Laboratory tools. The hydrogen atoms and Gasteiger charges for selected antiviral molecules were added and saved in .pdbqt format. The chemical names and structures of the compounds are shown in Table 1. The grid box dimensions for molecular docking were created around the key residues of the catalytic domain of the RdRP protein. Fifty different conformations were generated for each compound by using the Lamarckian GA. All the generated conformations of each lead antiviral molecule were analyzed in PyMol 2.1.1 and Discovery studio 2017 R2 software. Further, DINC 2.0 web server based on a metadocking algorithm (http://dinc.kavrakilab.org) was used to compute the binding affinities of the selected antiviral compounds with RdRP (Antunes et al., 2017). Further, HADDOCK was also used to perform molecular docking of Galidesvir, F3407-4105, F6523-2250, F6559-0746, BDG 33693278, BDG 33693315, and LAS 34156196 with RdRP (De Vries et al., 2010; Kumari et al., 2021). The protein–molecule interaction figures were prepared by using PyMOL (DeLano, 2002) and Maestro (Schrödinger Release 2020-3: Maestro, Schrödinger, LLC, New York, NY, 2020).
Chemical Information of the Potential Antiviral Molecules
2.5. Determination of molecular properties
The pkCSM web server (http://structure.bioc.cam.ac.uk/pkcsm) was used to compute the drug-like properties, including molecular weight, log P, number of rotatable bonds, hydrogen bond acceptors and donors (Lipinski's rule of five), aqueous solubility, skin permeability, human intestinal absorption, the volume of distribution, cytochrome P450, CYP2D6 inhibition, total clearance, Ames toxicity, and skin sensitization (Absorption, Distribution, Metabolism, Excretion and Toxicology [ADMET] properties) of the screened compounds (Lipinski, 2004; Pires et al., 2015).
2.6. MD simulation
The atomistic binding stability of protein along with ligands was investigated by using MDsimulation. MD was carried out by using GROMOS96 43a1 force field in GROMACS on NMRbox platform (van Gunsteren, 1996; Van Der Spoel et al., 2005; Maciejewski et al., 2017; Jairajpuri et al., 2021). The topology files of Galidesivir, F3407-4105, F6523-2250, F6559-0746, BDG 33693278, BDG 33693315, and LAS 34156196 were generated by PRODRG (Schüttelkopf and Van Aalten, 2004; Kesari et al., 2020). The systems were solvated in a triclinic box along with simple point charges. The volume of the box was 1395.37 nm3, and the minimum distance between edges of the box and protein was set at 1.0 nm. Overall charges on the systems were neutralized by additions of counterions (12 Na+) and minimized by 50,000 steps of descent algorithm, as previously done (Singh et al., 2018). The systems were equilibrated for constant number of particles, volume, and temperature (NVT) and constant number of particles, pressure, and temperature (NPT) for 1 ns as done by Dhankhar et al. (2020). During NVT and NPT, temperature (300°K) and pressure (1 bar) were maintained by Berendsen temperature coupling method and Parrinello-Rahman pressure coupling method (Parrinello and Rahman, 1981; Berendsen et al., 1984). The MD was run for 100 ns, and the generated trajectory files were used to analyze the results.
2.7. Molecular mechanics poisson-boltzmann surface area binding free energy
Binding free energy of RdRP-Galidesvir and RdRP-inhibitor(s) complexes was calculated by using g_mmpbsa (Kumari et al., 2014; Prasanth et al., 2020). A total of 2000 frames extracted from the last 20 ns (80–100 ns) of MD were used to estimate the Molecular Mechanics Poisson-Boltzmann Surface Area (MMPBSA) binding free energy of Galidesvir, F3407-4105, F6523-2250, F6559-0746, BDG 33693278, BDG 33693315, and LAS 34156196 with RdRP.
3. Results
3.1. Retrieval of three-dimensional structure of RdRP and MSA
The crystal structure of SARS-CoV-2 RdRP protein (PDB ID: 7C2K) was obtained from the RCSB PDB database and used as the target. The SARS-CoV-2 protein, RdRP (nsp12) comprised an N-terminal domain (1–249 a.a), an interface domain (250–365 a.a), and a right-hand polymerase domain (367–920 a.a). The active site of the polymerase domain is formed by seven polymerase motifs (A to G) and consists of crucial catalytic residues Asp618 (motif A) and Asp760, Asp761 (motif C), which are involved in a template and nucleotide binding and catalysis (Fig. 2) (Wang et al., 2020b).
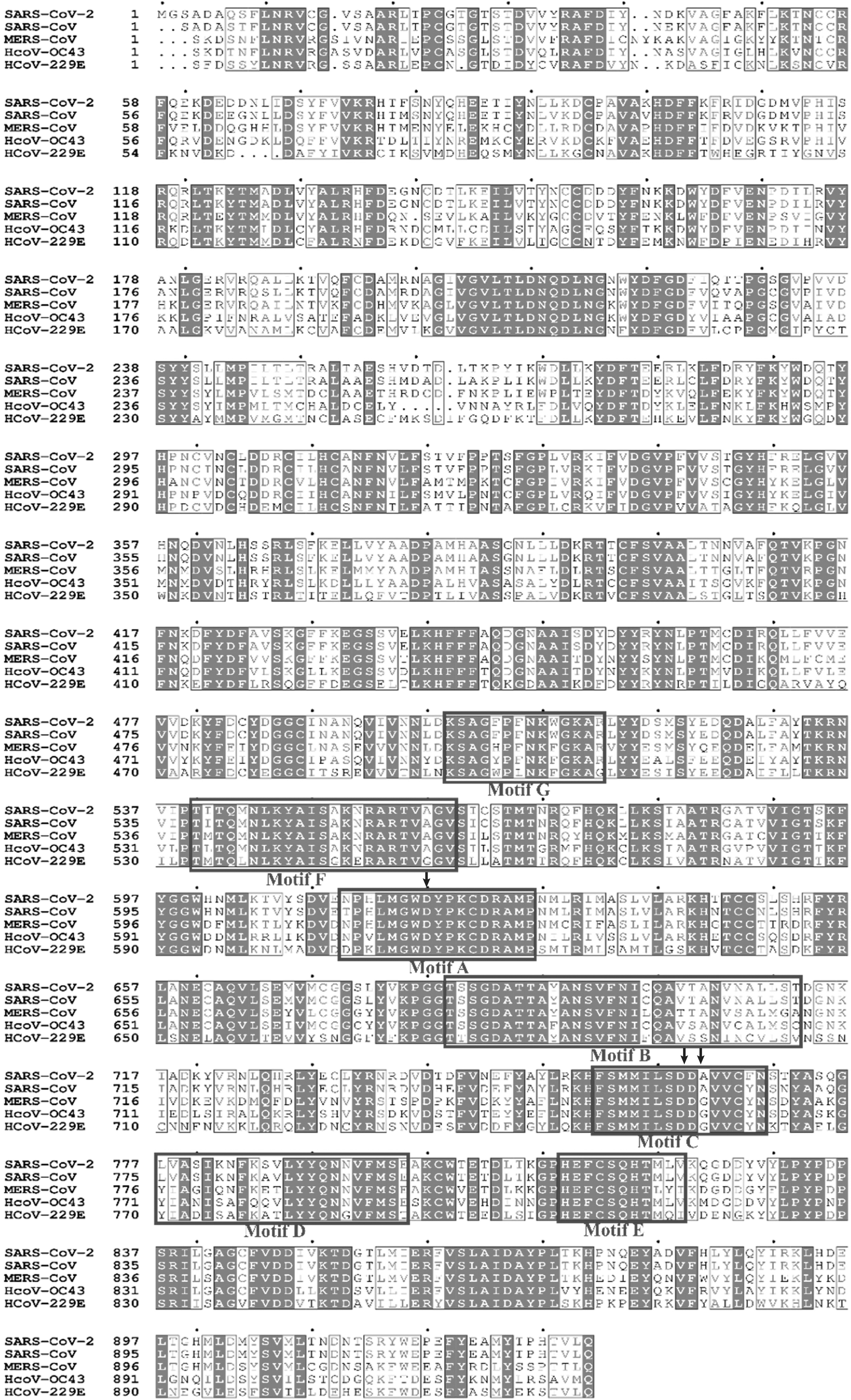
MSA of SARS-CoV-2 RdRP with its homologs. Polypeptide sequence of SARS-CoV-2 RdRP was aligned with those of its homologs from four other human beta coronaviruses, namely SARS-CoV, MERS-CoV, HCoV-OC43, and HCoV-229E. Clustal Ω was used to align these sequences, and the figure was generated by using ESPript 3. The conserved residues are represented in gray scale. Key residues, Asp618, Asp670, Asp671, are shown with arrow heads. HCoV, human coronavirus; MERS-CoV, middle east respiratory syndrome coronavirus; MSA, multiple sequence alignment.
3.2. Molecular docking of existing drug against RdRP of SARS-CoV-2
To identify the potent molecules against RdRP protein of SARS-CoV-2, six FDA-approved RdRP inhibitors were selected as positive controls. The binding of these positive controls at the active site of RdRP was verified (Fig. 3A and Table 2). These molecules perfectly fit into the active site of RdRP, engaging the conserved active residues (Asp760, Asp761) and getting stabilized by several favorable interactions, including hydrogen bonding and hydrophobic interactions. The binding energies of these positive controls were in the range of −3.67 to −5.59 kcalmol−1 with Favipiravir<Peniciclovir<Remdesivir<Sofosbuvir<Ribavirin<Galidesivir in that order (Fig. 3B and Table 2). Galidesivir has the highest binding affinity and is stabilized by the formation of conventional hydrogen bonds and hydrophobic interactions (Fig. 3C and Table 2). Therefore, we selected Galidesivir with binding energy −5.59 kcalmol−1, as the standard positive drug to be used for comparison with novel potential RdRP inhibitors from LC and ASINEX libraries.

Interaction of SARS-CoV-2 RdRP and existing inhibitors.
The Binding Affinities (kcalmol−1) of the Existing Drug Molecules with Active Site Residues of RNA-Dependent RNA Polymerase
3.3. Virtual screening of antiviral compounds
The antiviral molecules were virtually screened against RdRP by using the AutoDock Vina in PyRx 0.8, from LC and ASINEX libraries (Trott and Olson, 2010; Dallakyan and Olson, 2015). The downloaded antiviral molecules from both libraries were converted into .pdbqt format by using Open Babel and docked into the catalytic pocket of RdRP. The compounds had higher binding affinity (> −7.5 kcalmol−1) than Galidesivir. Moreover, these selected antiviral molecules from the library have a similar type of architecture, that is, an aromatic ring-like structure with a number of hydrogen acceptors and donors. These compounds also formed a stable protein–ligand complex through hydrogen bonds and hydrophobic interactions with Arg553, Thr556, Asp618, Tyr619, Lys621, Asp623, Arg624, Asp760, and Asp761 of RdRP.
3.4. Re-docking using GA of top hits
Molecular docking simulation was used to determine the binding affinity of the molecule against the target protein, RdRP. However, the standard error in the scoring function could lead to improper selection of the lead compound. Thus, re-docking was employed to eliminate false positives in the screening process. Therefore, all possible inhibitors from both libraries were re-docked separately against RdRP protein to discover the effective ligands and important atomic interactions between the protein–ligand complexes within the drug-binding cavity. The screened antiviral molecules were F3407-4105, F6523-2250, and F6559-0746 from LC library and BDG 33693278, BDG 33693315, and LAS 34156196 from ASINEX library.
The molecular docking study of all six screened antiviral molecules with RdRP revealed that all of them showed higher binding energy and inhibition constants than Galidesivir with RdRP. The obtained binding energies and inhibition constants are provided in Table 3. The analysis of docking results of screened molecules revealed a number of protein–ligand interactions, including hydrogen bond and hydrophobic interactions with the active site residues of RdRP, also detailed in Table 3. The selected antiviral molecules and positive control Galidesivir engaged the same residues from RdRP active site in their interactions with the protein. The result analysis of Galidesivir reveals the binding with a docking score of −5.59 kcalmol−1, and it was stabilized at the active site of RdRP by five conventional hydrogen bonds with Tyr619, Asp623, Asp760, and Asp761 (Fig. 3C). Galidesivir also formed hydrophobic interactions such as π-alkyl bonding, and π-donors with RdRP, as shown in Figure 3B. Further, analysis of screened compounds unveiled that the six antiviral compounds (F3407-4105, F6523-2250, and F6559-0746 from LC and BDG 33693278, BDG 33693315, and LAS 34156196 from ASINEX) had higher docking scores than Galidesivir, as enlisted in Table 3.
The Binding Affinities (kcalmol−1) of the Selected Antiviral Molecules with the Interacting Residues in RNA-Dependent RNA Polymerase Predicted from the Molecular Docking Studies
The molecule F3407-4105, from LC library, was able to establish four H-bonds with Tyr619, Asp623, and Asp760 (Fig. 4A). The complex of RdRP-F3407-4105 was also stabilized by hydrophobic interactions, including π-donors (with Tyr619, Cys622, and Ser759), π-anions (with Asp760 and Asp761), and π-alkyl (with Lys621 and Cys622), as shown in Figure 4D. The molecule F6523-2250 in complex with RdRP was able to form three H-bonds with Thr556, Lys621, and Asp760 of RdRP (Fig. 4B). Besides H-bonds, hydrophobic interactions including π-anions bonds (with Asp618, Asp623, and Asp761), π-alkyl bonds (with Lys621 and Arg624), π-donor bonds (with Tyr619), and π-cationic (with Arg553) were also observed for the stability of RdRP-F6523-2250 complex, as shown in Figure 4E. In another complex of RdRP-F6559-0746, it was observed that F6559-0746 formed five H-bonds with RdRP residues: Tyr619, Cys622, Thr687, Asn691, and Asp760 (Fig. 4C). This complex was also stabilized by π-anionic bonds (with Asp623 and Asp761), π-sulfur bonds (with Cys622), and π-sigma bonds (with Asp623), as shown in Figure 4F.

Compounds identified from Life Chemicals library against RdRp protein. Based on the stable conformations inferred from docking F3407-4105
Likewise, in the RdRP-BDG 33693278 complex, seven H-bonds were established by BDG 33693278 with Asp452, Arg553, Ala554, and Tyr619 (Fig. 5A) and other bonds, such as alkyl bond (with Cys622 and Arg555) and π-cationic bond (with Arg624), also provided stability to the RdRP-BDG 33693278 complex (Fig. 5D). Similarly, in the complex of RdRP with BDG 33693315, three conventional H-bonds were formed by BDG 33693315 with Tyr619, Cys622, and Asp761 of RdRP (Fig. 5B). Other than conventional H-bonds, the hydrophobic interactions such as π-anionic bonds (with Asp623 and Asp761), π-alkyl bonds (with Lys621 and Arg624), alkyl bonds (with Cys622), and carbon H-bonds (with Asp618, Tyr619, and Glu811) were involved in the stabilization of the complex, RdRP-BDG 33693315 (Fig. 5E). Another molecule, LAS 34156196, formed four conventional H-bonds with Asp761, Trp800, and Glu811 of RdRP in the RdRP-LAS 34156196 complex (Fig. 5C). Further, this complex was also stabilized by carbon H-bonds, ionic interactions, and π-interactions such as π-anionic, π-donor, and π-alkyl (Fig. 5F). HADDOCK docking results were also in accord with AutoDock results and showed a higher binding affinity of identified potent molecules than Gaidesvir (Table 4).

Compounds identified from ASINEX library against RdRp protein.
Molecular Docking Results of Galidesvir, F3407-4105, F6523-2250, F6559-0746, BDG 33693278, BDG 33693315, and LAS 34156196 with RNA-Dependent RNA Polymerase by HADDOCK
From these observations, it is evident that all the six identified have higher potential to engage active site residues in interactions than Galidesivir, thereby implicating higher RdRP inhibitory ability than the positive control.
3.5. Determination of molecular properties
To evaluate the drug likeness of the identified potential antiviral candidates, Lipinski's rule of five and ADMET tests were employed. The selected lead, having higher binding affinities than Galidesivir, successfully fulfilled the criteria for Lipinski's rule of five, including molecular weight, LogP, hydrogen bond donor, and acceptor (Table 5). The results showed that all the compounds mentioned earlier had a molecular weight of less than 500 Da, a LogP less than 5, and H-bond acceptors and donors of less than 10 and 5, respectively.
Different Physicochemical Properties of the Top-Scored Binding Affinity Molecules Fulfilling Lipinski's Rule of Five (Log –APS–#x1D443;–/APS– ≤ 5, Molecular Weight ≤500 Da, Number of Hydrogen Bond Acceptors ≤10, and Number of Hydrogen Bond Donors ≤5)
A promising ADMET profile of small molecules is essential in drug discovery. Thus, pharmacokinetic parameters such as aqueous solubility (log S), skin permeability coefficient (logKp), logBB, central nervous system (CNS) permeability, number of metabolic reactions, etc., and toxicity of molecules were evaluated and summarized in Table 6. The results indicated that all six compounds were soluble in water. The aqueous solubility (S) of a compound substantially affects its absorption and distribution properties. All compounds showed skin permeability and also exhibited absorbability by the human intestine. In addition, they were able to penetrate the blood–brain barrier into the CNS after oral administration. CYP enzymes, various CYP450 substrates, and inhibitors play a fundamental role in the metabolism of the drug. Further, metabolic analysis revealed that none of these compounds were substrates of the CYP2D6 inhibitor. Toxicity assessment showed that all compounds are potentially non-toxic to humans, although some of the predicted values might reflect low tolerance of the body.
The Absorption, Distribution, Metabolism, Excretion, and Toxicology Profile of the Potential Antiviral Molecules
BBB, blood–brain barrier; CNS, central nervous system.
3.6. MD simulation
MD, which is a vital computerized method to study the dynamic stability of protein–ligand complexes, was carried out to check the stability of RdRP-Galidesvir and RdRP-inhibitor(s) complexes, as done by Dalal et al. (2019). The MD trajectories were retrieved to calculate the Root mean square deviation (RMSD), radius of gyration (Rg), solvent accessible surface area (SASA), hydrogen bond, and MMPBSA binding energy.
The dynamics stability of ligands with RdRP was described by RMSD, as it evaluates the motion in the protein's backbone during the MD. The RMSD results of the RdRP-Galidesvir complex attained equilibrium around 48 ns, whereas RdRP-inhibitor(s) complexes were equilibrated around 29 ns and remained stable up to the end of molecular simulation (Fig. 6A). The average RMSD values of RdRP-Galidesivir, RdRP-F3407-4105, RdRP-F6523-2250, RdRP-F6559-0746, RdRP-BDG 33693278, RdRP-BDG 33693315, and RdRP-LAS 34156196 complexes were 0.42 ± 0.04, 0.36 ± 0.02, 0.37 ± 0.03, 0.39 ± 0.03, 0.37 ± 0.02, 0.38 ± 0.03, and 0.40 ± 0.04 nm, respectively, as shown in Table 7. From Figure 6B and Table 7, it can be clearly seen that all the identified compounds had lesser RMSD than Galidesvir. Overall, RMSD results displayed that RdRP-inhibitor(s) complexes were more stable than the RdRP-Galidesvir complex.
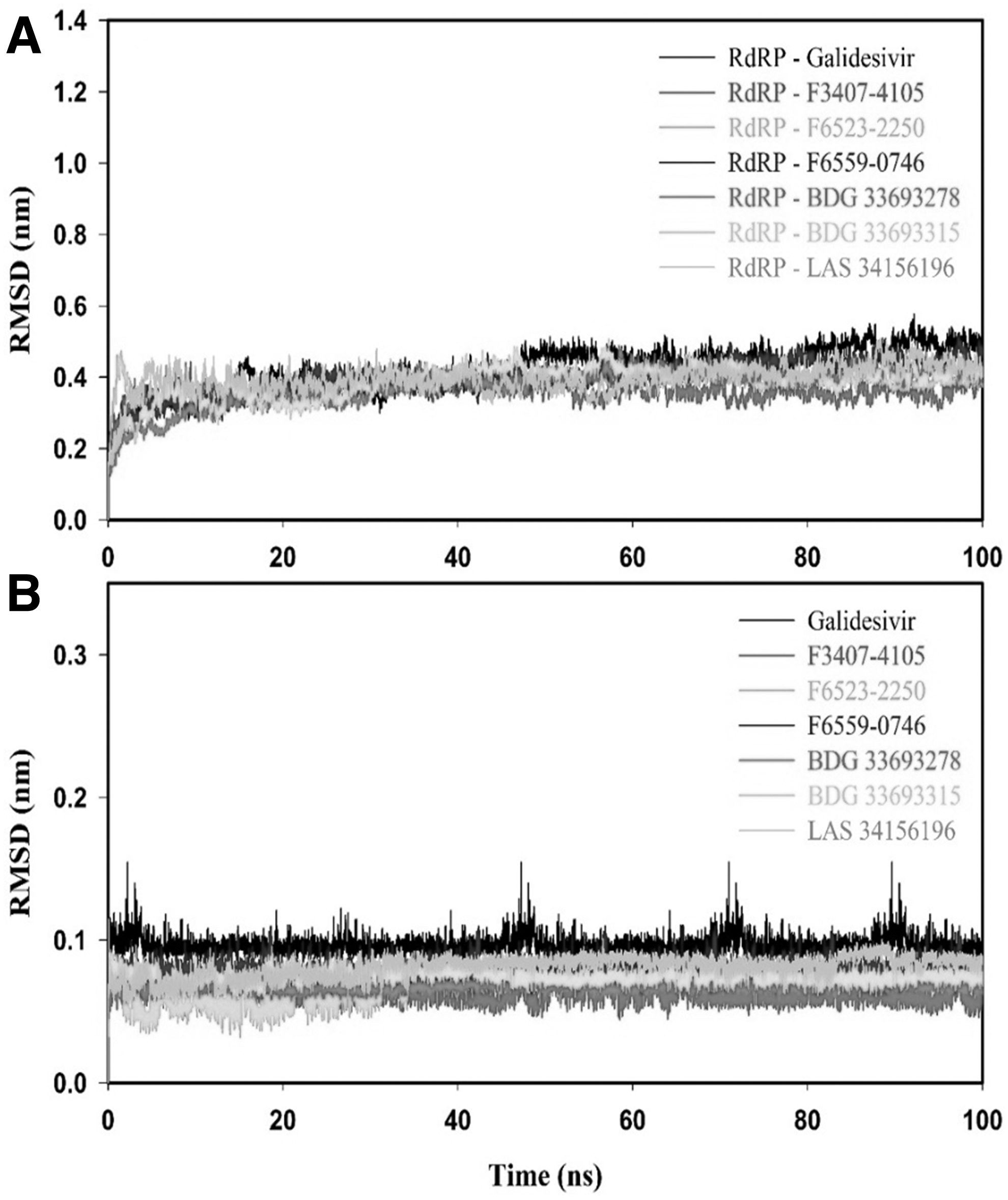
RMSD plots protein-ligand complexes
Average of Molecular Docking Results (Root Mean Square Deviation for Protein and Ligand, Rg, and Solvent Accessible Surface Area) of RNA-Dependent RNA Polymerase-Galidesvir, RdRP-F3407-4105, RdRP-F6523-2250, RdRP-F6559-0746, RdRP-BDG 33693278, RdRP-BDG 33693315, and RdRP-LAS 34156196 Complex for the Time Period of 100 ns
RMSD, root mean square deviation; SASA, solvent accessible surface area.
The Rg of RdRP-Galidesvir and RdRP-inhibitor(s) complex was determined to define the protein–ligand complex's compactness during the MD simulation. We have plotted the Rg values versus time scale for all the complexes and it revealed that RdRP-inhibitor(s) complexes were more compact and stable as compared with the RdRP-Galidesvir complex (Fig. 7). The average Rg values for RdRP-Galidesivir, RdRP-F3407-4105, RdRP-F6523-2250, RdRP-F6559-0746, RdRP-BDG 33693278, RdRP-BDG 33693315, and RdRP-LAS 34156196 complexes were 3.12 ± 0.02, 3.05 ± 0.02, 3.06 ± 0.02, 3.09 ± 0.02, 3.05 ± 0.02, 3.08 ± 0.01, and 3.10 ± 0.02 nm, respectively, as shown in Table 7. SASA results indicated that the RdRP-inhibitor(s) complex was more stable than the RdRP-Galidesvir complex (Fig. 8). Average SASA values of RdRP-inhibitor(s) complexes were in the range of 348.06 ± 8.28 to 358.94 ± 8.06 nm2, which was lesser than 361.12 ± 7.41 nm2, as shown in Table 7.
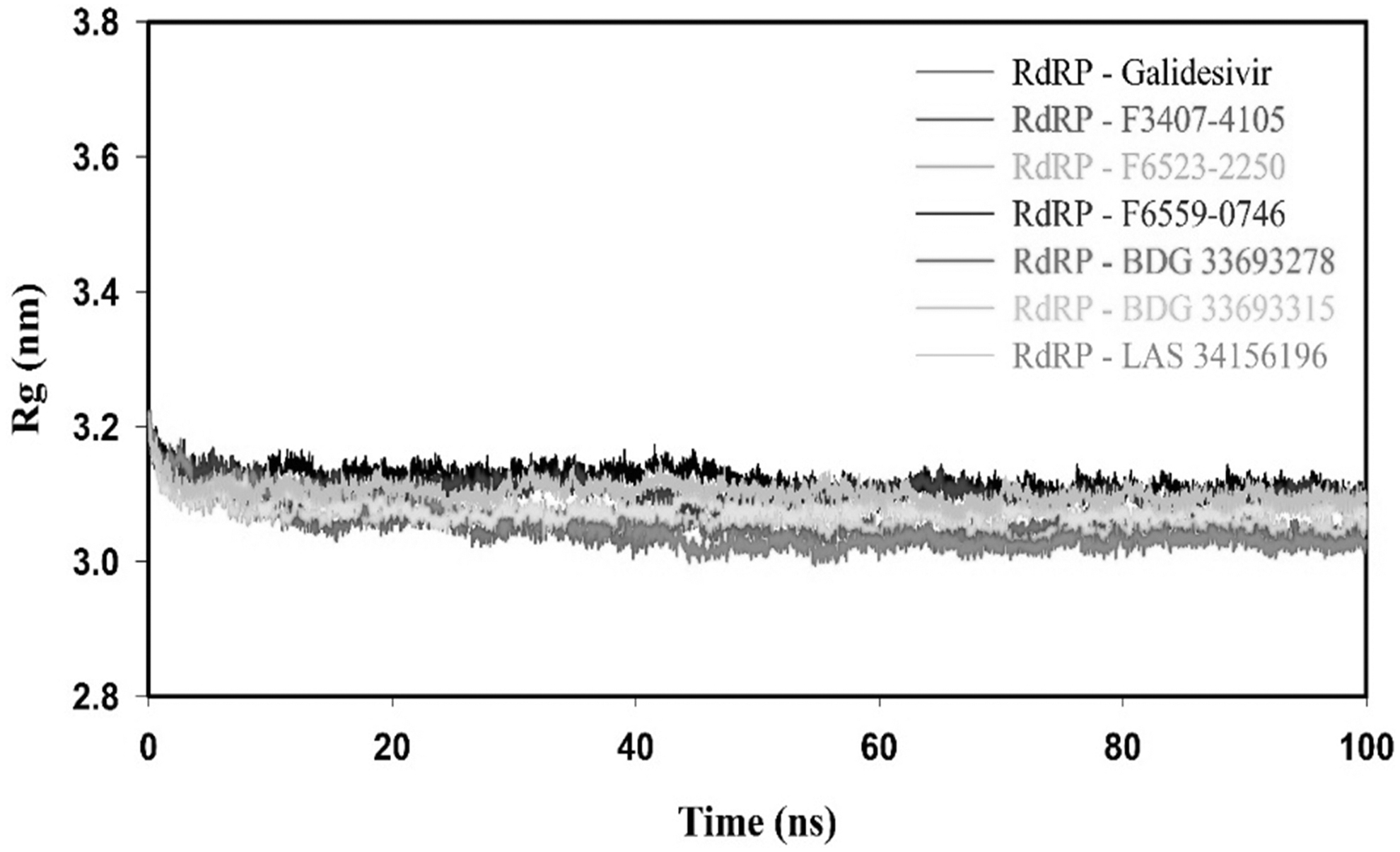
Rg plot of RdRP-Galidesvir and RdRP-inhibitor(s) complexes for the duration of 100 ns of MD at 300°K. Rg, radius of gyration.

SASA results for RdRP-ligand(s) complexes. SASA profile for RdRP-Galidesvir, RdRP-F3407-4105, RdRP-F6523-2250, RdRP-F6559-0746, RdRP-BDG 33693278, RdRP-BDG 33693315, and RdRP-LAS 34156196 during the molecular simulation of 100 ns. SASA, solvent accessible surface area.
Hydrogen bond plays a vital role in the stabilization of protein–ligand complexes. Therefore, we monitored the intraprotein and intermolecular hydrogen bonds to analyze the RdRP-Galidesvir and RdRP-inhibitor(s) complex (Fig. 9). The RdRP-inhibitor(s) complexes showed higher intraprotein hydrogen bonds than the RdRP-Galidesvir complex (Fig. 9A). RdRP-Galidesivir, RdRP-F3407-4105, RdRP-F6523-2250, RdRP-F6559-0746, RdRP-BDG 33693278, RdRP-BDG 33693315, and RdRP-LAS 34156196 complexes had an average of intraprotein hydrogens 725.9 ± 16.3, 738.5 ± 16.2, 730.9 ± 16.4, 730.7 ± 16.3, 738.9 ± 16.7, 736.6 ± 17.3, and 727.8 ± 15.9, respectively (Table 7). The RdRP-inhibitor(s) complexes had higher intermolecular hydrogen bonds as compared with the RdRP-Galidesvir complex (Fig. 9A). Overall, hydrogen bond results indicated that the binding of identified molecules formed higher stable RdRP-inhibitor(s) complex than the RdRP-Galidesvir complex.
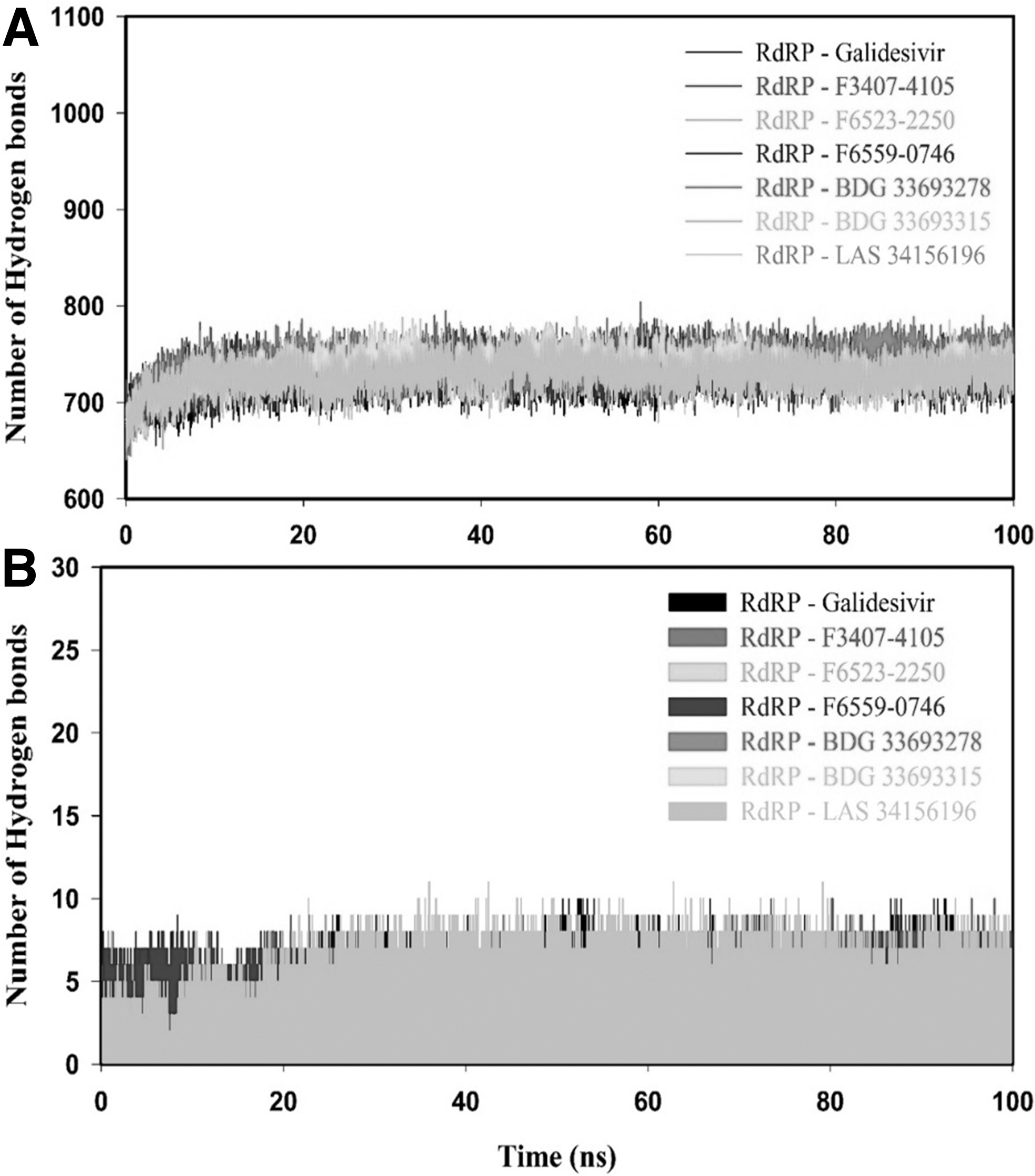
Hydrogen bond analysis of RdRP–ligand(s) complexes.
3.7. MMPBSA binding free energy
The binding free energy of Galidesvir, F3407-4105, F6523-2250, F6559-0746, BDG 33693278, BDG 33693315, and LAS 34156196 with RdRP was determined from the last 20 ns (80–100 ns) of MD. The MMPBSA binding free energy of RdRP-Galidesvir, RdRP-F3407-4105, RdRP-F6523-2250, RdRP-F6559-0746, RdRP-BDG 33693278, RdRP-BDG 33693315, and RdRP-LAS 34156196 was −168.19 ± 3.79, −223.54 ± 2.64, −213.41 ± 3.23, −211.19 ± 3.66, −224.12 ± 3.38, −215.69 ± 3.18, and −206.96 ± 3.85 kJmol−1, as shown in Table 8. The MMPBSA binding free energy results clearly showed that binding of identified molecules with RdRP resulted in the formation of a lower energy stable RdRP-inhibitor(s) complex than the RdRP-Galidesvir complex.
Molecular Mechanics Poisson-Boltzmann Surface Area Binding Free (kJmol−1) of RNA-Dependent RNA Polymerase-Galidesvir, RdRP-F3407-4105, RdRP-F6523-2250, RdRP-F6559-0746, RdRP-BDG 33693278, RdRP-BDG 33693315, and RdRP-LAS 34156196 Complexes Calculated from the Last 20 ns (80–100 ns) of Molecular Dynamics
4. Discussion
COVID-19 currently poses serious survival challenges to the worldwide population. This zoonotic disease is caused by SARS-CoV-2, which belongs to the betacoronavirus genus of coronaviridae family of RNA viruses (Wu et al., 2020b; Zhou et al., 2020). HCoV are known to affect global health, and in the past two decades, SARS-CoV-2 is the third HCoV after SARS-CoV and MERS-CoV that caused severe problems in humans across the world (Lee et al., 2003). Till now, there is no effective medicine and treatment for this disease. Therefore, new antiviral drugs for SARS-CoV-2 are imperative.
In this study, we have carried out the computational screening by using the RdRP protein of SARS-CoV-2 to identify potential antiviral molecules. RdRP is a well-known multi-functional protein and it plays a vital role in the replication of a virus. As RdRP is conserved across the positive-sense RNA viruses, it is a highly effective target. Recently, several studies using computational drug design approaches have been performed to identify FDA-approved drugs as potential SARS-CoV-2 inhibitors (Iftikhar et al., 2020; Kandwal and Fayne, 2020). Elfiky et al. reported the binding of five approved antiviral drugs, including Galidesivir, Remdesivir, Tenofovir, Sofosbuvir, and Ribavirin, to RdRP of SARS-CoV-2 (Elfiky, 2020b). Further, the structural studies on RdRP of SARS-CoV-2 revealed that nucleotide analogs, including Remdesivir, Favipiravir, Ribavirin, Galidesivir, and Sofosbuvir, strongly bind with the residues (Asp618, Asp760, and Asp761) of the active site to block the viral RNA synthesis and this study suggests that the outcomes of docking are reliable (Elfiky, 2020b; Gao et al., 2020; Khazeei Tabari et al., 2020; Yin et al., 2020).
In the current study, we have also docked Galidesivir, Ribavirin, Sofosbuvir, Remdesivir, Peniciclovir, and Favipiravir with RdRP and the docking scores were −5.59, −5.32, −5.27, −4.85, −4.44, and −3.67 kcalmol−1, respectively. The results showed that out of all the FDA-approved antiviral drugs, Galidesivir showed the highest binding affinity and stability through hydrogen and hydrophobic interactions at the active site of RdRP. Thus, Galidesivir was considered as a reference molecule to identify the potent novel candidates by virtual screening.
A total of 10,327 antiviral molecules from two libraries (LC and ASINEX) were subjected to virtual screening by using the reported crystal structure of RdRP. Out of the top 30 screened molecules, six (F3407-4105, F6523-2250, F6559-0746, BDG 33693278, BDG 33693315, and LAS 34156196) interacted at the active site of RdRP with binding energies higher than Galidesivir. The molecular docking results for all six potential compounds indicated binding of these compounds to the active site of RdRP with higher binding energy (−8.4 to −9.0 kcalmol−1) than Galidesivir (−5.59 kcalmol−1).
The binding affinity computed by the DINC server also indicated that all six antiviral molecules bound at the active site of RdRP with an affinity higher than Galidesivir. These compounds and Galidesivir formed a stable protein–ligand complex through a network of molecular interactions, including hydrogen bonds and the hydrophobic interactions with the key residues (Asp618, Asp760, and Asp761) of RdRP. The molecules were analyzed on the basis of binding energy and molecular interaction with crucial residues of the active and catalytic site. The overall results revealed that the identified molecules from both the libraries showed more stable H-bonds, salt bridges, and ionic interactions as compared with FDA-approved drugs. Some predicted tolerable doses with negative values suggest that the body might have low forbearance for these drugs. However, these are just predictive values that are considered to be helpful in deciding the explorative doses for initial preclinical studies. Moreover, the six antiviral molecules, under consideration complied well with Lipinski's rule of five and fulfilled the ADMET properties, suggesting that these molecules could be potential inhibitors of SARS-CoV-2 RdRP.
MD was employed to understand the stability of RdRP-Galidesvir and RdRP-inhibitor(s) complexes. The lesser RMSD of RdRP-inhibitor(s) complexes indicated that the binding of the identified compounds with RdRP results in the formation of a higher stable RdRP-inhibitor(s) complex than the RdRP-Galidesvir complex. Rg results revealed that RdRP-inhibitor(s) complexes were more compact than the RdRP-Galidesvir complex. SASA and hydrogen bond analysis also supported the outcomes of RMSD and Rg results that the binding of identified molecules with RdRP tend to form the higher stable RdRP-inhibitor(s) complex than the RdRP-Galidesvir complex. The MMPBSA results confirmed that the RdRP-inhibitor(s) complex had a higher binding affinity (−206.96 ± 3.85 to −224.12 ± 3.38 kJmol−1) than the RdRP-Galidesvir complex (−168.19 ± 3.79 kJmol−1). Overall, MD and MMPBSA analysis concluded that all the identified molecules can form lower energy stable RdRP-inhibitor(s) complexes than the RdRP-Galidesvir complex.
5. Conclusion
Due to the current COVID-19 pandemic, there is an urgent need for a new antiviral drug. This study focused on blocking the replication machinery of SARS-CoV-2 by targeting RdRP protein, which helps in the replication of the SARS-CoV-2 virus.
A potent drug should interact with the key residues of the active site of the polymerase and it blocks the positive sense strand RNA binding, which serves as a template. Blocking the active site with these antiviral drugs can inhibit the synthesis of the new viral genome, and consequently, block the formation of mature virus particles. In this study, all the identified candidates from LC and ASINEX libraries have shown stable interactions with the key residues of RdRP, with binding energies higher than Galidesivir. These compounds also satisfied the parameters for a potent inhibitor. The MD analysis also revealed that the identified molecules along with RdRP were more stable as compared with the RdRP-Galidesvir complex. The MMPBSA results confirmed that RdRP-inhibitor(s) complexes were lower energy complex than the RdRP-Galidesvir complex.
These results indicated that the identified molecules are potent antivirals that can be used to inhibit the activity of RdRP. Thus, these molecules warrant for further in vitro and preclinical analyses toward developing into marketable drugs.
Footnotes
Authors' Contributions
P.D. and V.K. conceived the basic idea of the study, performed virtual screening and molecular docking studies, and analyzed the data. V.D. conducted the molecular dynamics studies. P.D. wrote the first draft. V.K. reviewd and edited the article.
Acknowledgments
V.K. acknowledges the scholarship received from the Council of Scientific and Industrial Research, Govt. of India. This study made use of NMRbox: National Center for Biomolecular NMR Data Processing and Analysis, a Biomedical Technology Research Resource (BTRR), which is supported by NIH grant no. P41GM111135 (NIGMS).
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This study was not supported by any funding.