
Abstract
Deep neural networks (DNNs) have been recently proposed for quartet tree phylogeny estimation. Here, we present a study evaluating recently trained DNNs in comparison to a collection of standard phylogeny estimation methods on a heterogeneous collection of datasets simulated under the same models that were used to train the DNNs, and also under similar conditions but with higher rates of evolution. Our study shows that using DNNs with quartet amalgamation is less accurate than several standard phylogeny estimation methods we explore (e.g., maximum likelihood and maximum parsimony). We further find that simple standard phylogeny estimation methods match or improve on DNNs for quartet accuracy, especially, but not exclusively, when used in a global manner (i.e., the tree on the full dataset is computed and then the induced quartet trees are extracted from the full tree). Thus, our study provides evidence that a major challenge impacting the utility of current DNNs for phylogeny estimation is their restriction to estimating quartet trees that must subsequently be combined into a tree on the full dataset. In contrast, global methods (i.e., those that estimate trees from the full set of sequences) are able to benefit from taxon sampling, and hence have higher accuracy on large datasets.
1. INTRODUCTION
One of the basic challenges in statistical phylogeny estimation occurs when sequences evolve under processes that violate assumptions of the inferential statistical model. For example, most phylogeny estimation methods are based on sequence evolution models, such as the Generalized Time Reversible (GTR) (Tavaré, 1986) model for nucleotide evolution, that assume that all the sites within the sequences evolve i.i.d. (identically and independently) down a model tree. In turn, the sequence evolution models make several simplifying assumptions (such as stationarity, homogeneity, and time-reversibility, jointly referred to as “SRH”) about sequence evolution to ensure statistical identifiability (i.e., that the model tree is uniquely identified by the probability distribution it defines on site patterns) and computational feasibility (i.e., so that tree estimation can be performed by using acceptable efforts).
However, these are unrealistic assumptions, as compositional heterogeneity (indicative of changed substitution rate matrices) across the tree has been observed in many biological datasets [e.g., see discussion in Jermiin et al. (2004)]. Indeed, recent years have shown an increasing awareness of the impact of these violations of the model assumptions on phylogeny estimation (Jermiin et al., 2004; Kolaczkowski and Thornton, 2008; Duchêne et al., 2017; Naser-Khdour et al., 2019; White and Braun, 2019; Crotty et al., 2020), leading Naser-Khdour et al. (2019) to conclude:
the extent and effects of model violation in phylogenetics may be substantial […] further effort in developing models that do not require SRH assumptions could lead to large improvements in the accuracy of phylogenomic inference.
To address the challenge of model misspecification, several investigators have developed parameter-rich models (Steel, 1994; Crotty et al., 2020) and methods for estimating trees under these models; for example, IQ-TREE 2 (Minh et al., 2020) enables estimation under the GHOST (Crotty et al., 2020) model, which incorporates substantial heterogeneity across the tree and across sites. However, two challenges appear with an increased number of parameters: The computational cost increases, and there is a danger of overfitting (and hence reduction of accuracy).
An alternative approach that has recently begun to be explored is the use of deep neural networks (DNNs), which are machine-learning models that can be trained on datasets and then used to classify new input datasets. The DNNs have been developed for computing phylogenies on datasets of four sequences (i.e., quartet tree estimation), so that each DNN is trained on sets of four sequences with known true quartet trees, and then used to estimate quartet trees from new sets of four sequences (Suvorov et al., 2020; Zou et al., 2020).
This approach has the benefit of allowing the training to be done under very heterogeneous conditions, including ones that significantly violate standard model assumptions. Further, although the training may be computationally intensive, once the training is complete quartet tree estimation is very fast.
Other applications of DNNs to phylogeny estimation are less direct. For example, Bhattacharjee and Bayzid (2020) used machine learning to develop techniques to impute the missing entries in a distance matrix, and so enable the reconstruction of trees from partial distance matrices. Abadi et al. (2020) suggested a machine-learning framework, ModelTeller, for phylogenetic model selection. Leuchtenberger et al. (2020) designed and trained DNNs to distinguish quartet trees exhibiting the properties of the Felsenstein Zone (Felsenstein, 1978) or the Farris Zone (Siddall, 1998), using both simulated and empirical datasets.
This approach does not directly construct quartet trees but does provide guidance to the user in the subsequent choice of method for the phylogeny estimation method for the dataset. Recently, Jiang et al. (2021) developed DEPP, a deep learning-based approach for phylogenetic placement. Even more recently, Azouri et al. (2021) explored the use of a deep-learning algorithm to narrow the tree search space to achieve faster convergence of the maximum likelihood (ML) score.
In this study, we focus on the use of DNNs presented in Suvorov et al. (2020) and Zou et al. (2020), which operate by estimating quartet trees (i.e., unrooted binary trees on just four leaves). To extend the use of these DNNs to estimate larger trees, the quartet trees computed by the DNN must be combined together into a tree on the full dataset, a process referred to as “quartet amalgamation.”
However, optimization problems for quartet amalgamation, such as maximizing the number of satisfied quartet trees, are NP-hard (Jiang et al., 2001), which necessitates the use of heuristic approaches. Nevertheless, many quartet amalgamation methods have been developed (Strimmer and Von Haeseler, 1996; Ranwez and Gascuel, 2001; Snir et al., 2008; Reaz et al., 2014), with Quartets Max Cut (QMC) now the leading quartet amalgamation method.
We specifically examine two questions: (1) How accurate are the quartet trees estimated by DNNs in comparison to other standard methods and (2) how accurate are trees computed by DNNs on just slightly larger trees with 20 sequences, in comparison to standard methods? We use the trained DNNs from Zou et al. (2020) for the quartet tree estimators, and QMC for the quartet amalgamation method, thus producing a two-phase approach that we refer to as DNN+QMC.
We simulate sequences using the simulator developed by Zou et al., using the same basic model conditions as they used for training and then extending the set of model conditions to include a wider range of branch lengths. Significantly, we find that DNNs are less accurate than standard methods for both problems, even when considered on the same data on which they were trained. Indeed, in our study, the commonly used ML methods under standard i.i.d. sequence evolution models are more accurate than these DNNs for quartet tree estimation and more accurate than DNN+QMC methods at estimating 20-taxon trees, even though the datasets we examine have evolved under substantial violations of the model assumptions. Further, even neighbor joining (NJ) (Saitou and Nei, 1987) used with p-distances (i.e., normalized Hamming distances) is more accurate than DNN+QMC methods.
Thus, these DNNs do not even match the accuracy of basic phylogeny estimation methods. We provide some insights into why this is likely to be true, focusing on taxon sampling and its beneficial effects in phylogeny estimation. Overall, this study provides a cautionary note about the use of DNNs for phylogenetic gene tree estimation, while indicating some possible directions for their use in phylogenomics.
2. MATERIALS AND METHODS
2.1. Overview
We explore the accuracy of the three DNNs used in Zou et al. (2020). Each of these was trained on a different training set, and they are named after their training datasets; these are DNN1, DNN2, and DNN3. We compare these DNNs with standard phylogeny estimation methods [ML, maximum parsimony (MP) (Foulds and Graham, 1982), NJ, and unweighted pair group method with arithmetic mean (UPGMA) (Sokal and Michener, 1958)] with respect to tree topology accuracy on a range of model conditions. We performed two experiments to understand the performance of DNNs in comparison to standard phylogeny estimation methods. Experiment 1 evaluates the topological accuracy of estimated 20-leaf trees and quartet trees on simulated 200 aa sequence datasets. Experiment 2 performs the same evaluation but on longer sequences (1000 aa). See Appendix A1 for the detailed commands.
2.2. Datasets
We simulated datasets with 20 gap-free amino acid sequences using the evosimz simulator from Zou et al. (2020). Three of the model conditions use the same parameter values as the training datasets used in Zou et al. (2020) to train DNN1, DNN2, and DNN3, but we restricted the trees to 20 leaves [while Zou et al. (2020) explored a larger range of tree sizes]. The basic model conditions are called Training1, Training2, and Training3. For the main quartet tree estimation experiments reported in Zou et al. (2020) (i.e., the “nolba” conditions, which do not involve long-branch attraction), quartet tree error rates were extremely low: never more than 7%, and most analyses were much better (e.g., MP never had more than 5% error).
This level of quartet tree accuracy is an indication that the tree estimation problem is unusually easy under these test model conditions, and hence atypical of most phylogenetic problems. Therefore, in our study, we modified the simulator parameter settings to produce more challenging conditions. We achieved this in two ways. First, we modified the branch length distributions to increase the rate of evolution. Second, we noted that the main experiments they report were based on sequence lengths drawn from a uniform distribution [100, 3000), so that the median length is about 1550.
Since sequence length has a large impact on phylogeny estimation accuracy, we examined two sequence lengths: 200 aa (a length that is close to typical lengths of single proteins) and 1000 aa [a length that would be much less frequently observed, but still not as unrealistic as the lengths evaluated in Zou et al. (2020)].
We created 12 model conditions, with 4 conditions per basic model condition. Specifically, for a given basic model condition (Training1, Training2, Training3), we include the original model condition, and we also include three other conditions defined by modifying the branch length distribution to provide higher upper bounds on the branch lengths, to make for more challenging datasets. For each of the 12 model conditions, 20 trees were generated with 20 leaves each, and gap-free amino acid sequences evolved down the tree under heterogeneous substitution processes that include heterotachy.
With the exception of branch lengths, we set all the numeric simulation parameters identically as for the associated model conditions drawn from Zou et al. (2020). We extended the range of branch lengths as follows: We set the upper bound of the branch length distribution to X times the original upper bound, where X was set to
We also modified the lower bound on the branch length distributions for the Training1-based and Training3-based model conditions. The model parameters for the different model conditions are provided in Supplementary Table S1, and associated empirical statistics of these model conditions are provided in Supplementary Table S3; see also Supplementary Table S2 for information on how to interpret branch lengths.
In Experiment 1, we used all 12 model conditions, and we explored methods on sequences with 200 aa. For Experiment 2, we used 9 of the 12 model conditions (omitting only the three hardest model conditions with the highest rate of evolution) and examined performance on sequences with 1000 aa; thus, Experiment 2 reflects performance on very long protein sequence alignments, a condition that is likely to be much less common than Experiment 1.
2.3. Phylogeny estimation methods
We use a collection of phylogeny estimation methods in this study, starting with the trained DNNs from Zou et al. (2020) and including some standard phylogeny estimation methods.
The DNNs classifiers. We used DNN1 (Epoch 588), DNN2 (Epoch 1272), and DNN3 (Epoch 1098), obtained from Zou et al. (2020), reflecting the final training state achieved by the authors. DNN1 was trained on quartet trees generated by the first training set (a superset of Training1), DNN2 was trained on quartet trees generated by the second training set (a superset of Training2), and DNN3 was trained on quartet trees generated by the third training set (a superset of Training3). To compute trees on more than four sequences using DNNs, we combine the quartet trees into a tree on the full dataset using QMC (Snir and Rao, 2008), in default mode. We refer to this two-phase approach as DNN+QMC (i.e., DNN1+QMC, DNN2+QMC, and DNN3+QMC).
Standard methods. For ML analyses, we use the Linux version of IQ-TREE v. 2.0.5 (Minh et al., 2020) in three ways: under the WAG (Whelan and Goldman, 2001) model, under the GHOST (Crotty et al., 2020) model, and using ModelFinder (Kalyaanamoorthy et al., 2017) to select the best fitting model according to a BIC criterion. For the MP, NJ, and UPGMA analyses, we use PAUP* v. 4.0a (Swofford, 2002), running these methods in default mode; in particular, both NJ and UPGMA are run by using uncorrected distances (i.e., p-distances, which is the fraction of the sequence length where the two sequences have different amino acids).
Local and global methods for quartet tree estimation. The “local” approach for quartet tree estimation is used on four sequences at a time, and it estimates quartet trees by using only the local information and no additional information. For example, the local version of NJ for quartet tree estimation takes four sequences, computes the
In contrast, the “global” approach operates as follows. Given a set of
Every phylogeny estimation method we explore, thus, can be used in either a local or global way to estimate quartet trees. The description given earlier for the local and global variants of NJ makes it clear as to how these terms are used for MP, UPGMA, and ML under three models. For the DNNs, the local version is the normal usage of the DNN (i.e., the input is a set of four aligned gap-free sequences, and the output is the quartet tree it computes). The global version of the DNNs is obtained by running DNN+QMC followed by extraction of the tree on the specified quartet.
2.4. Criteria
We evaluate methods with respect to tree topology error rates, computed in two different ways: quartet tree error rates and bipartition error rates. For quartet tree error, we record the percentage of the quartet trees computed that are incorrectly reconstructed. For bipartition error (applied for the 20-leaf datasets), we report the Robinson and Foulds (1981) error rates, defined as follows (see the Appendix A1 for more details). Every edge in a tree defines a bipartition on the leaf set, so that the Robinson-Foulds (RF) distance between two trees is the number of bipartitions that appear in one tree but not in the other. Finally, the RF error rate for an estimated tree on n leaves is its RF distance to the true (model) tree divided by
3. RESULTS
3.1. Experiment 1: Accuracy on datasets with 200 aa
Here, we explore tree estimation accuracy on datasets with sequences having 200 aa. Experiment 1(a) examines the accuracy of the 20-leaf trees we compute, and Experiment 1(b) examines quartet tree accuracy.
3.1.1. Experiment 1(a): Accuracy on 20-leaf trees
For each training set and method, the most accurate results are obtained on the base model, and error rates then increase as the rate of evolution increases (Table 1 and Fig. 1). These results are expected and reflect the impact of the rate of evolution on phylogenetic estimation difficulty [e.g., see Liu et al. (2011)]. In addition, error rates are lowest on the Training1-based model conditions and highest on Training3-based conditions, so the Training2-based conditions are of intermediate difficulty.
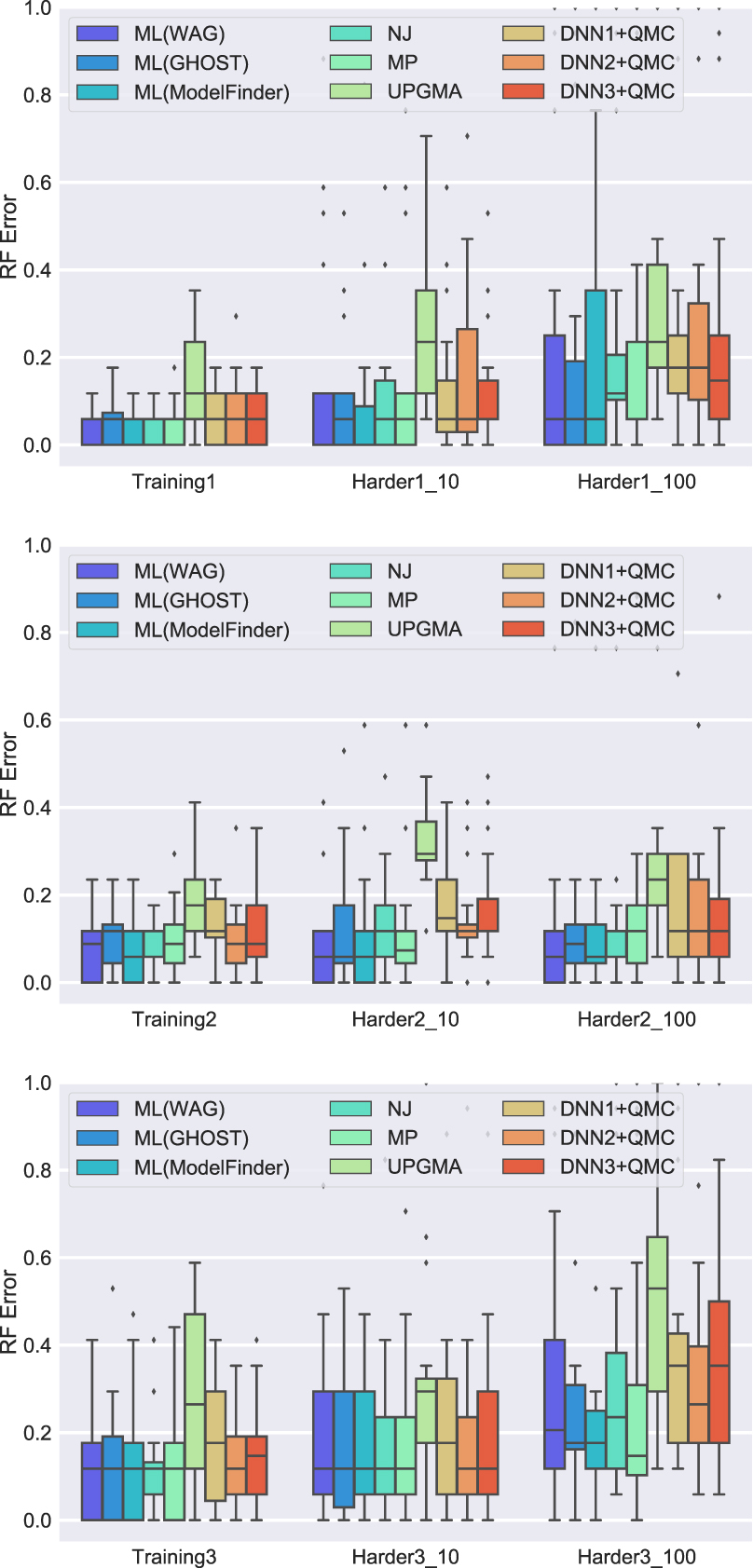
Experiment 1: Global tree error on 20-leaf datasets with 200 aa.
Experiment 1: Tree Error (Median Robinson-Foulds Rates) of 20-Sequence Datasets with 200 aa Estimated Under the Different Model Conditions (20 Replicates)
Boldface values indicate top place.
NJ and UPGMA are run by using uncorrected distances. The ML methods are performed by using IQ-TREE 2 in default mode, under the indicated models.
MF, ModelFinder; MP, maximum parsimony; ML, maximum likelihood; NJ, neighbor joining; QMC, Quartets MaxCut; UPGMA, unweighted pair group method with arithmetic mean.
Across all 12 model conditions, UPGMA has the worst accuracy, and the standard phylogeny estimation methods (NJ, ML, and MP) have the best accuracy. Thus, the DNNs only improve on UPGMA. With UPGMA set aside, the relative performance between methods depends on the specific model condition. The accuracy of ML under IQ-TREE 2 depends on the choice of the model, with an advantage to trees estimated under models selected by ModelFinder (see Supplementary Table S4). The difference between NJ, MP, and the ML methods is generally small, but when there is a difference, it tends to favor the ML methods. Interestingly, MP is the most accurate method of all on the Harder3_1000 condition.
The comparison between the DNNs depends on the model condition, but the differences tend to be small. On the model conditions derived from Training1 and Training2, the three DNNs (DNN1+QMC, DNN2+QMC, DNN3+QMC) are all about the same in terms of accuracy (with perhaps a slight advantage to DNN2+QMC). On the model conditions derived from Training3, DNN2+QMC is slightly better than DNN1+QMC and DNN3+QMC. Thus, overall we see that DNN2+QMC seems to have a small advantage over DNN1+QMC and DNN3+QMC.
It is also worth noting how often a given method comes in first place (or ties for first place) in this experiment. ML using Model Finder comes in first place 11 out of 12 times, followed by MP (7 times), ML under the WAG model (6), ML under Ghost and NJ (both at 6 times), and then DNN2+QMC (1). The remaining DNN+QMC methods never come in first place in any model condition.
In sum, therefore, the DNN+QMC methods do not match the accuracy of ML under simple models. Further, very simple methods, such as NJ on p-distances and MP, are much more accurate than the DNN+QMC methods.
3.1.2. Experiment 1(b): Accuracy of quartet tree methods
Performance of local quartet tree estimation methods. Table 2 shows quartet tree error rates for the local methods under the 12 model conditions. As expected, the error rates increase for all methods as the rate of evolution increases; however, many of the other trends are surprising. For example, one of the most striking findings in this study is that NJ is the best method, with the lowest median error for all model conditions. This is surprising, given that NJ is used with the simplest distance calculation, p-distances (percentage of aligned sites that are different).
Experiment 1(B): Error Rates (Medians) of Quartet Trees Estimated Using Local Quartet Methods on Datasets with 200 aa Under Different Model Conditions (20 Replicates)
Boldface values indicate top place.
NJ and UPGMA are run by using uncorrected distances. ML methods are performed by using IQ-TREE 2 in default mode, under the indicated models.
Further, NJ has a consistent advantage over the DNNs, which can be substantial (e.g., Harder1_100, Harder2_100, and Harder3_100). In contrast, UPGMA has the worst accuracy, indicating that the advantage of NJ is not because distance-based methods are generally at an advantage.
Also intriguing is that ML under simpler models is more accurate than ML under more complex models, despite the fact that the simulation protocol introduced very substantial heterogeneity. For example, ML(WAG) generally has better accuracy than ML under GHOST (the most complex model) and under the model selected by ModelFinder.
Another interesting finding is that MP has better accuracy than ML(GHOST) for every model condition, and it is better than ML (MF) for 9 of the 12 model conditions. Indeed, only ML under the simplest model condition, WAG, is more accurate than MP (and even for this, MP improves on ML(WAG) on four model conditions, ties on one condition, and is never more than 2% worse).
The three DNNs have similar accuracy across all the model conditions (and no single DNN outperforms the others) and never tie for the first place. In essence, therefore, the DNNs are not particularly good as local quartet tree estimators, and very simple methods are strictly better.
Performance of global quartet tree estimation methods. In comparing methods as global quartet tree estimation, we also see substantial differences. The key observation is that quartet trees computed using DNN+QMC methods are not as accurate as quartet trees computed using most other global methods, with the only exception being UPGMA, which has the worst accuracy (Table 3). Further, across the 12 model conditions, ML(WAG) came in the first place in eight conditions, NJ on p-distances and ML(MF) came in the first place in six conditions, MP came in the first place in five conditions, and the three DNN+QMC methods came in the first place in one to three model conditions. Thus, DNN+QMC methods are not anywhere near the best performing global quartet tree methods.
Experiment 1B: Median Quartet Error on the 20 Replicates of Each Model Condition Using a Global Quartet Estimation Approach on Datasets with 200 aa.
Boldface values indicate top place.
NJ and UPGMA are run by using uncorrected distances. ML methods are performed by using IQ-TREE 2 in default mode, under the indicated models.
Comparing local and global quartet tree methods. To better understand the limitations of DNNs as quartet tree methods, we now examine the difference in accuracy between quartet trees computed using local methods and quartet trees computed using global methods. We compare the best local ML method [i.e., ML(WAG)], the three DNNs, NJ, and MP; however, we omit UPGMA because of its generally poor accuracy seen in the previous experiments.
For every model condition and for every method, it is always better to use the global version of the method instead of the local version of the method (see Fig. 2 for one model condition and Supplementary Figs. S1–S3 for the other models). The difference between local and global quartet tree error rates depends on the model condition (smaller differences between these error rates under the slower rate of evolution than under higher rates of evolution) and method. Even under the lowest rate of evolution, the differences are not small: The difference in accuracy ranges from 3% (for NJ) to about 12% (for ML under the GHOST model). Under the highest rate of evolution, the difference for ML methods under all models ranges up to 27% and up to 21% for the DNN methods.
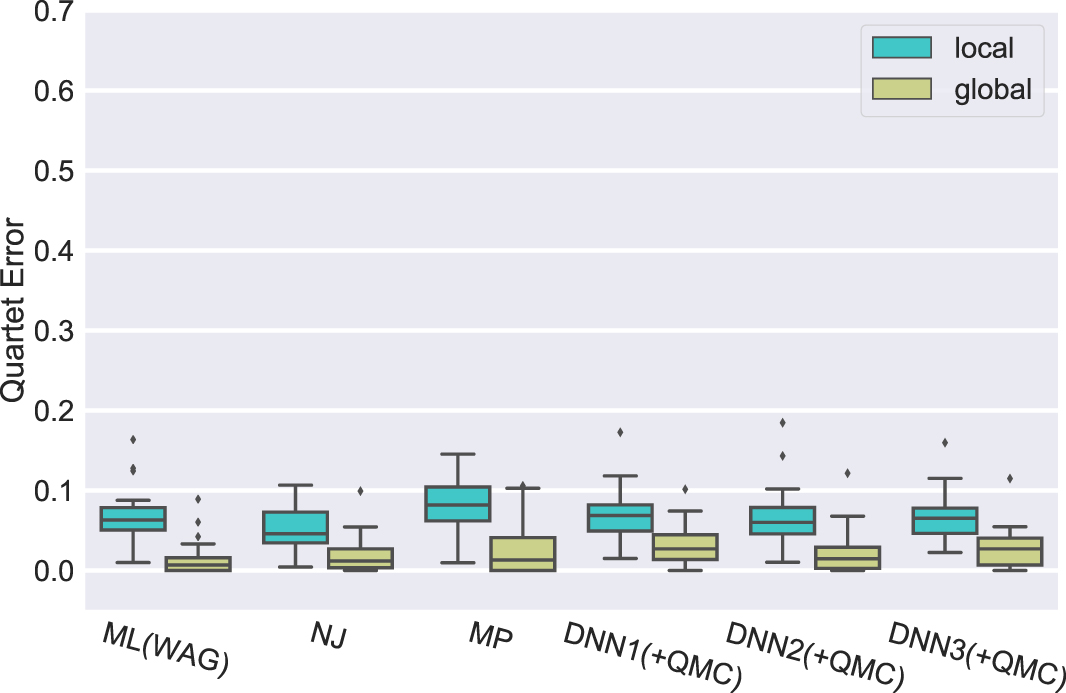
Experiment 1: Comparison of quartet tree error of local and global versions of each tree estimation method on Training2 datasets with 200 aa. The local approach estimates quartet trees directly on 4-sequence alignments, whereas the global approach estimates quartets by first computing the 20-leaf tree and then each individual quartet is extracted.
3.2. Experiment 2: Accuracy on long sequences
Here, we explore results on 1000 aa sequences. Examining RF error rates on the 20-leaf trees (Table 4), we see that error rates for all methods are much lower than on shorter sequences (200 aa), and that the differences between methods are reduced. Indeed, for three of the nine model conditions all methods recover the true tree exactly, and there are only two model conditions where any method has worse than 6% RF error. Thus, the model conditions we explore in Experiment 2 are much easier than the model conditions explored in Experiment 1.
Experiment 2: Tree Error (Median Robinson-Foulds Rates) of 20-Leaf Trees Estimated Under the Different Model Conditions (20 Replicates) on Long Sequences (1000 aa)
Boldface values indicate top place.
NJ and UPGMA are run by using uncorrected distances. ML methods are performed by using IQ-TREE 2 in default mode, under the indicated models.
However, even under these easier conditions, there are still clear differences in RF error between methods. For example, four methods (the three ML methods and MP) come in the first place in eight of the nine model conditions, and the remaining methods (NJ and the three DNN+QMC methods) come in the first place between four and six times. The three ML methods and MP also never have error rates above 6% for any model condition, whereas NJ has error rates between 9% and 12% for two model conditions, and the three DNN+QMC methods have error rates between 15% and 18% for the hardest model condition.
Thus, although these are easier model conditions and differences between the top methods are reduced, the DNN+QMC methods are still not competitive for accuracy with the other methods.
Comparing these methods as local quartet tree methods (Table 5) is also relevant. Here, we see the DNNs producing very good quartet trees, each coming in the first place in five of the eight model conditions; only ML(WAG) has this same good performance (and the remaining methods come in the first place at most three times). Thus, as local quartet tree methods, when analyzing long sequences, the DNNs can provide good value.
Experiment 2: Error Rates (Medians) of Quartet Trees Estimated Using Local Quartet Methods Under the Different Model Conditions (20 Replicates) on Long Sequences (1000 aa)
Boldface values indicate top place.
NJ is run by using uncorrected distances. ML methods are performed by using IQ-TREE 2 in default mode, under the indicated models.
Interestingly, we see different relative performance when comparing methods for global quartet tree accuracy (Table 6): The DNN+QMC methods come in the first place in only four out of the nine conditions, and each of the other methods comes in at least five times. The best in terms of global quartet tree accuracy is ML(WAG), which is best in eight of the nine conditions, followed by NJ, which is best in seven out of nine conditions, and then by MP, which is best in six out of nine conditions. Thus, as global quartet tree methods, the DNN+QMC approach clearly lags behind the other methods, even for these much easier model conditions.
Experiment 2: Error Rates (Medians) of Quartet Trees Estimated Using Global Quartet Estimation Under the Different Model Conditions (20 Replicates) on Long Sequences (1000 aa)
Boldface values indicate top place.
NJ is run is using uncorrected distances. ML methods are performed by using IQ-TREE 2 in default mode, under the indicated models.
4. DISCUSSION
We begin with a summary of the observations in this study. We examined methods for tree construction on either four-leaf datasets or on 20-leaf datasets, and with two sequence lengths and different rates of evolution. When constructing 20-leaf trees, we see very clear advantages to several standard tree estimation methods (notably, ML and MP) compared with DNN+QMC methods. These differences were especially pronounced for our first experiment where we examined accuracy on datasets with 200 aa per sequence, but also evident in our second experiment where we examined performance with longer sequences (1000 aa per sequence).
When used to construct quartet trees, however, the relative accuracy depended on the sequence length. For short sequences, the best method was NJ based on p-distances (i.e., uncorrected distances), an exceedingly simple method, and the DNNs were not particularly accurate. Interestingly, for long sequences (1000 aa), the DNNs were tied with ML (WAG) as the best local quartet tree estimators. Clearly, therefore, sequence length impacts the relative accuracy of DNNs and other methods, used as local quartet tree methods.
However, the most accurate quartet trees were computed by constructing trees on the full set of 20 sequences using one of the standard approaches (i.e., NJ, MP, and ML) and then inducing the quartet trees; further, the global version of quartet tree estimation was always more accurate than the local version. We observed that DNN+QMC methods are also more accurate at estimating quartet trees than the DNNs themselves, showing that the QMC amalgamation method is able, to some extent, to correct errors in the estimating quartet trees through this amalgamation step. Even so, the final quartet tree accuracy of DNN+QMC methods does not match the quartet tree accuracy of the global methods.
Comparison to Zou et al. (2020). The trends reported in Zou et al. (2020) may at first glance seem inconsistent with what we have observed. Although we mainly focused on the accuracy of methods on large (20-leaf) trees, there also seem to be differences between trends we observed for local quartet methods and the trends they observed (i.e., they concluded that DNNs have the best accuracy on simulated datasets, and are “overall comparable” to the existing methods on biological datasets).
An important consideration to note is that the majority of simulation conditions Zou et al. (2020) explored were all very easy due to long sequence lengths and reduced rates of evolution. However, even for the model conditions we explored that had the same rates of evolution as their main model conditions (i.e., Training1, Training2, and Training3), our results did not show their DNNs having the best accuracy, even as local quartet methods—instead we saw NJ having the best accuracy. How do we reconcile these differences in observations?
A better understanding of their DNNs on their own test data can be seen by examining results shown in their Figure 2b, which includes a wider range of sequence lengths. This figure demonstrates that sequence length has a large impact on the accuracy of their DNNs (and on other methods), and it also reveals that their DNNs were less accurate than NJ on the shortest sequences they examine (100–200 aa). Thus, the relative performance between their DNNs and NJ seems to depend on the sequence length.
Further, our results on the model conditions with low rates of evolution (i.e., our Training1, Training2, and Training3 model conditions) match their results for similar conditions. Thus, there are no difference in trends observed on simulated data in Zou et al. (2020) and our study when restricting attention to model conditions with the same properties.
It is also helpful to examine the local quartet error rates obtained by their DNNs and the other phylogeny estimation methods on five biological datasets, as reported in Table 2 of Zou et al. (2020) (summarized in our Supplementary Table S5). Analyses of the Mammals (mitochondrial) dataset produced very high error local quartet rates for all methods (ranging from 0.57 to 0.97 error), making comparisons based on this dataset uninformative and potentially misleading. Therefore, we restrict our attention to the remaining four datasets. On the red fluorescent protein dataset, NJ and MP were the best approaches, followed by RAxML.
Then, for the Mammalian genes dataset, NJ performed the best by far, followed by the ML approaches (RAxML and PhyML). On the Mammals (concatenated) dataset, MP and RAxML gave the best results, followed closely by the other non-DNN approaches, and with all the DNNs performing worst. Similarly, for the Plants (concatenated) dataset, NJ was the best approach, followed by all other methods, except for DNN3, which performed the worst.
Overall, therefore, the results reported in Zou et al. (2020) reveal that the DNNs were clearly less reliable than NJ and most other methods on the biological datasets as local quartet tree methods.
These trends suggest that these DNNs can be accurate (and competitive with other methods) for local quartet tree estimation given relatively long sequences that have evolved under sufficiently low rates of evolution but have not been shown in our experiments to provide comparable accuracy to the better standard methods for “large” tree estimation (measured using RF error or global quartet error).
It is possible that under other conditions, such as long branch attraction or high levels of heterotachy, DNN methods might be as accurate, or perhaps even more accurate, than standard methods, but the inferior performance of these DNN methods on biological datasets does suggest that these conditions are unlikely to be very general.
Why are DNN+QMC methods not competitive for large-tree estimation? Our study showed that DNN+QMC methods were not as accurate as simple methods for estimating 20-sequence trees; here, we examine the possible explanations for this trend.
The accuracy of these DNN+QMC methods depends on both the accuracy of the quartet trees computed using the DNNs and also the ability of QMC, the quartet amalgamation method we selected, to combine these quartet trees into an accurate tree on the full dataset. Our study clearly shows that the quartet trees, estimated independently of each other (i.e., “local methods”), are less accurate than quartet trees induced from a tree estimated on the full dataset (i.e., “global methods”).
This trend holds true for all the standard phylogeny estimation methods we explored, including NJ, ML, and MP. We also noted that QMC, used in conjunction with the DNNs, was able to improve the quartet score, but that this approach to quartet tree estimation was not as accurate as the truly global approach to estimating quartet trees. This seems to be an inherent limitation of DNN+QMC methods.
One might then ask whether QMC itself is a limiting factor, and that substantially better quartet amalgamation methods might exist and yield improved accuracy compared with standard methods. Although QMC is generally regarded as the best of the currently available quartet amalgamation methods, we explored the use of Quartet Puzzling (Strimmer and Von Haeseler, 1996) [the same method used in Zou et al. (2020)] to combine quartet trees computed using DNNs.
Our study found that Quartet Puzzling produced highly unresolved and much less accurate 20-leaf trees than QMC (see Supplementary Table S6). Given QMCs superiority to Quartet Puzzling and the general reception of QMC as a leading quartet amalgamation method, it seems unlikely that better amalgamation methods are available at this time.
Thus, based on our study, we hypothesize that any method that is used to construct a tree in this two-stage approach (i.e., by first computing quartet trees independently and then merging them with a quartet amalgamation method) is unlikely to be as accurate as a good global method. This hypothesis is based on much prior literature, which has shown that big trees can, in many cases, be easier to estimate with high accuracy than small trees [e.g., see the example in Hillis (1996) and subsequent discussion in Hillis (1998); Pagel and Meade (2008); Zwickl and Hillis (2002); Nabhan and Sarkar (2012)]. This, we posit, is likely to be the main limitation of using any two-stage technique that uses DNNs to estimate small trees (here, four-leaf trees) and then amalgamation methods to combine the smaller trees.
Impact of model complexity. A very interesting trend we observed is that ML under simple models often produces more accurate trees than ML under complex models. For example, on the datasets with 200 aa, ML(GHOST) was not as accurate as ML(WAG), despite GHOST being a more complex model (and most likely a better fit to the simulation model) and WAG being an extremely simple model. The advantage of ML(WAG) over ML(GHOST) is also present on the longer sequences when they are used as local quartet tree estimation methods but disappears when they are used as global methods. We conjecture that this may be due to overfitting, since the advantage to the simpler model is greatest when the total amount of data is the smallest.
Related to this, we note again the very high accuracy of MP compared with the ML-based methods, when used as a local quartet tree estimation on short sequences (200 aa): MP had better accuracy than ML(GHOST) for every model condition, was better than ML(MF) for 9 of the 12 model conditions, and was only less accurate than ML(WAG) (and even there it was very close in accuracy). These trends may be related to the equivalence between MP and ML under the no-common mechanism model, as proven in Tuffley and Steel (1997), and the relative accuracy of ML and MP on datasets simulated under heterogeneous models, as provided in Kolaczkowski and Thornton (2004).
5. CONCLUSIONS
The DNNs can be used as quartet tree estimators, and—when combined with quartet amalgamation methods, such as QMC—they can be used to construct larger trees. However, in our study, they were not as accurate at estimating 20-leaf trees as many standard phylogeny estimation methods, including MP, NJ, and ML under simple models of evolution. The failure of DNNs to provide good accuracy that is competitive with even simple methods is noteworthy and important to understand.
Our study shows that the advantage of standard methods (e.g., ML under simple models) over DNN+QMC methods is due mainly to the benefits inherent in being able to estimate the entire tree at once rather than through a two-stage approach that first estimates quartet trees (i.e., local quartet estimation) and then combines the quartet trees into a tree on the full dataset. The limitations of these two-stages approaches compared with global methods have been previously noted in other studies [e.g., St. John et al. (2003)], and these are closely related to the well-known benefits produced by dense taxon sampling.
One way to address this limitation is to design DNNs to estimate much larger trees (e.g., 10-leaf trees rather than 4-leaf trees). However, such an approach would only have limited success, since the estimation of much larger trees would still require amalgamation methods (called “supertree methods” when the input trees are not just quartet trees). Further, taxon sampling would still benefit global methods over local methods, even if the local methods were computing 10-leaf trees. Finally, training a DNN requires a large volume of representative datasets, a challenge that is clearly already a problem for training classifiers of 4-leaf trees.
Since the number of 10-leaf trees is already more than 1,000,000, training DNNs to classify 10-taxon trees would likely be prohibitively difficult. Thus, we predict that trying to address the limitations of this two-stage approach by constructing trees on larger subsets is unlikely to be generally successful.
Given the observed dependency on sequence length, it is possible that the DNN+QMC approach might be best suited to species tree estimation, which is based on multi-locus datasets and in some cases on large portions of whole genomes. However, multi-locus datasets evolve under an array of processes, including incomplete lineage sorting, gene duplication and loss, and gene flow, that results in different loci evolving under different tree topologies (Maddison, 1997); Degnan and Rosenberg, 2009).
Thus, to enable DNNs to be useful on multi-locus datasets, they would need to be trained on data that evolve under complex models that reflect these genome-scale processes, rather than under the models that assume that all sites evolve down a single tree topology. Although this would increase the training complexity, such an approach might well have better results, as a result of having longer sequences and also potentially more biological training data (i.e., true gene trees are rarely known, but true species trees may be reliably known for some sets of four species).
However, since standard methods can be statistically inconsistent in the presence of gene tree heterogeneity (Roch and Steel, 2015), the new DNNs would then need to be compared with phylogenomic species tree estimation methods that explicitly address gene tree heterogeneity [e.g., Heled and Drummond (2009); Mirarab et al. (2014); Richards and Kubatko (2020); Smith et al. (2020)], as these have been shown to provide improved accuracy compared with standard methods and also have strong theoretical guarantees.
However, although the use of DNNs for directly constructing phylogenies has not yet succeeded in matching the accuracy of existing methods (and the challenges to doing this seem very formidable), as discussed in Section 1, DNNs can be used in other ways to inform phylogeny estimation. Thus, although our study suggests substantial limitations for this way of using DNNs in phylogeny estimation methods (i.e., by constructing small trees using DNNs and then combining them into larger trees), there are alternative uses for DNNs that could be highly informative and beneficial in the estimation of phylogenies under realistically complex evolutionary scenarios.
DATA AND CODE AVAILABILITY
The python source code for these experiments is available at https://gitlab.engr.illinois.edu/gmartin6/simulating_quartets. The datasets used in this study are available at https://doi.org/10.13012/B2IDB-8921156_V1.
Footnotes
ACKNOWLEDGMENTS
The authors thank Z. Zou and J. Zhang for assistance in understanding and using their codes.
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
This work was supported in part by NSF grants 1513629 and 1458652 to T.W. This research is part of the Blue Waters sustained-petascale computing project, which is supported by the National Science Foundation (awards OCI-0725070 and ACI-1238993), the State of Illinois, and as of December 2019, the National Geospatial-Intelligence Agency. Blue Waters is a joint effort of the University of Illinois at Urbana-Champaign and its National Center for Supercomputing Applications.
Appendix
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.