
Abstract
Microbes can be found almost everywhere in the world. They are not isolated, but rather interact with each other and establish connections with their living environments. Studying these interactions is essential to an understanding of the organization and complex interplay of microbial communities, as well as the structure and dynamics of various ecosystems. A widely used approach toward this objective involves the inference of microbiome interaction networks. However, owing to the compositional, high-dimensional, sparse, and heterogeneous nature of observed microbial data, applying network inference methods to estimate their associations is challenging. In addition, external environmental interference and biological concerns also make it more difficult to deal with the network inference. In this article, we provide a comprehensive review of emerging microbiome interaction network inference methods. According to various research targets, estimated networks are divided into four main categories: correlation networks, conditional correlation networks, mixture networks, and differential networks. Their assumptions, high-level ideas, advantages, as well as limitations, are presented in this review. Since real microbial interactions can be complex and dynamic, no unifying method has, to date, captured all the aspects of interest. In addition, we discuss the challenges now confronting current microbial interaction study and future prospects. Finally, we point out several feasible directions of microbial network inference analysis and highlight that future research requires the joint promotion of statistical computation methods and experimental techniques.
1. Introduction
Microbes exist widely in the biosphere, from fertile soil to warm spring water and from hard rocks to thin atmosphere, and even in extreme environments, such as high-temperature deep-sea liquid vents, rocky crusts in the depths of the earth, and cold poles (Huber et al., 2007; Pikuta et al., 2007). Microbes play a vital role in the biochemical cycle of the earth. In the ecological cycle of the biosphere, microbes act not only as producers, but also as the main decomposers of organic matter, demonstrating their importance as a force driving the biochemical cycle (Falkowski et al., 2008; Madsen, 2011).
Massine numbers of microbes live in the human body, and their total number is estimated to be 10 times that of human cells (Micah et al., 2007). They are distributed in various parts of the human body, such as skin, mouth, and intestines. Among them, the intestine has the largest number and variety of microbes (Filyk and Osborne, 2016). Microbes often exist in the form of communities, and they affect their own environment through synergy, which, in turn, affects the composition and structure of the microbial community.
The genomes of these microbes constantly interact with the human genome to regulate the metabolism of the host, which is closely related to human health. In recent years, many studies have emerged to explore the relationship between microbes and human health. For example, intestinal microbes are closely related to obesity (Turnbaugh et al., 2009), diabetes (Qin et al., 2012), high blood pressure (Holmes et al., 2008), cardiovascular diseases (Wang et al., 2011), AIDS (Lozupone et al., 2013), and cancer (Schwabe and Jobin, 2013). Meanwhile, people's eating habits and lifestyles will also affect the structure of intestinal flora (Wu et al., 2011). The Human Microbiome Project initiated by the American National Institutes of Health uses metagenomics to study the relationship among human body surface, microbial flora, and human health (Turnbaugh et al., 2007; Huttenhower et al., 2012; Methé et al., 2012).
The microbiome refers to the collection of all microbial species and their genetic information and functions that exist in a specific environment. It includes not only interactions among microbes in the environment, but also the interactions between microbes and other species and even environments. In recent years, with the rapid development of sequencing technology, researchers can extract genomic information of all microbes from microbial communities in the natural environment. More and more novel bacterial species have been discovered, and microbial sequencing data have grown exponentially (Proal et al., 2017).
Such sequencing makes it easier to study the correlations among microbial communities and between microbes and hosts. For example, 16S rRNA sequencing is a widely used high-throughput sequencing technology for microbiome studies. The 16S rRNA gene is the most common genetic marker to identify microbes (Wooley et al., 2010), and its advantages are mainly reflected in three aspects. First, the 16S rRNA gene exists in all microbes and has the same function in most organisms, except for eukaryotic cells.
Second, the length of 16S rRNA gene can reach 1500 bp, which can provide enough variation to identify the types of microbes (Janda and Abbott, 2007). Third, 16S rRNA sequences have highly conserved regions among different species, which allow us to use universal primers to sequence multiple species at the same time and distinguish the type of microbes based on the given hypervariable regions (Chakravorty et al., 2007).
Compared with other sequencing technologies, 16S rRNA sequencing technology also provides fast sequencing speed and low sequencing cost, which are also important factors in its widespread application in microbiome research. In the specific sequencing process, the variable regions of the 16S rRNA gene sequence are first amplified and sequenced, and then the sequences with a similarity of 95%, 97%, or 99% are grouped into one operational taxonomic unit (OTU); the OTU count is then used as a proxy variable for potential microbial abundance (Schloss, 2010; Morgan and Huttenhower, 2012). Further, grouping 16S rRNA gene sequences to obtain OTUs can be achieved in three ways.
The first is to cluster similar sequences by using an unsupervised clustering algorithm (Schloss et al., 2009), and the second is to use a phylogenetic tree that integrates mutation and evolution information (Hamady et al., 2010). The third method is to use the supervised learning model obtained from the labeled training data to determine the OTU type corresponding to the sequences (Wang et al., 2007). This method is also applied to whole-genome shotgun sequencing. To control the experimental deviation caused, for example, by sequencing depth or biological sample size, the total count in the sample is typically used to normalize the count of each OTU (Ozsolak and Milos, 2011; Ni et al., 2013; Fernandes et al., 2014).
These normalized data only reflect the relative abundance information of each OTU. In this way, microbiome sequencing data typically consist of compositional data (Kurtz et al., 2015).
A wide range of interactions takes place among microbes coexisting in nature. These interactions are vital to the earth's biochemical cycle and have a profound impact on human health and disease. Based on their influence on members of the microbial community, the various interactions among microbes can be divided into three types: beneficial, neutral, and harmful (Lidicker, 1979; Faust and Raes, 2012; Zengler and Zaramela, 2018). Beneficial interactions include commensalism and mutualism. The former means that the interaction is beneficial to one microbe but has no effect on other microbes.
The latter indicates that microbes coexist and benefit from each other. Neutral interaction represents the absence of any effect on both microbes. Harmful interactions are mainly divided into four situations: amenalism, competition, parasitism, and predation. Amenalism means that the connection is harmful to one microbe but has no effect on other microbes. Competition implies that all microbes involved in the interaction are at a disadvantage owing to the existence of other microbes. Parasitism occurs when microbes live off of their host, benefiting themselves at the expense of their hosts.
Predation means that one microbe feeds on another microbe. Together, these interactions constitute a complex ecological network that determines the structure, composition, and function of the microbial community in the ecosystem.
In microbiology and systems biology research, probabilistic graph models are typically used to characterize the interactions between various microbes in a microbial community (Jordan, 2003; Friedman, 2004; Whittaker, 2009; Cao et al., 2019). We can regard each kind of microbe as a vertex. If an interaction occurs between two microbes, we can connect their corresponding vertices to form an edge. In this way, we can get a network model describing the interactive relationship between microbes, namely the microbial interaction network.
Further, we can determine the strength of the interaction between microbes by assigning weights to edges such that the greater the absolute value of the weight, the stronger the interaction. A positive weight indicates beneficial interaction, and a negative weight indicates harmful interaction. Zero weight means neutral interaction. In the probabilistic graphical model, the absolute abundance of each microbe is regarded as a random variable, making the absolute abundance of all microbes in the microbial community a random vector. Microbiome studies are concerned with how the interaction network corresponds to each component in this random vector.
The goal of microbiome compositional data network analysis is to make statistical inferences about the interaction network corresponding to their unobserved absolute abundance by utilizing the observed relative abundance data of various microbes. Such interactions are often inferred by estimating the covariance matrix. In detail, interactions can be divided into those that are direct and those that are indirect. Indirect interaction may be mediated between two microbes, or it may be transmitted through a third microbe.
Direct interactions require no mediator or third factor and are more intrinsic. Since the two correlative estimation methods noted earlier do not make such distinctions, conditional correlation network inference methods that can estimate direct interactions have gained much attention. In addition, the interaction network between microbes is not constant for a long time, but changes as the external environment changes, such as the health status of the host, environmental conditions, or genetic conditions.
Hence, research on differential networks and mixture networks has recently become an important topic in microbiome studies. Mixture networks account for the heterogeneity of microbiome data and generalize the single underlying biological network into multiple networks. Differential networks focus on the dynamics of interaction networks and explore interactions from the aspect of comparison.
Previously, several researchers have reviewed the inference methods of microbiome networks, but they mainly focused on correlation networks (Cardona et al., 2016; Weiss et al., 2016; Dohlman and Shen, 2019). In contrast, in this article, we review some of the statistical calculation methods developed in recent years based on all four main network situations: correlation networks, conditional correlation networks, mixture networks, and differential networks. We summarize the high-level idea of these methods and discuss their model assumptions and scope of applications. We also provide some comments and prospects for future development of algorithms. Our article is organized as follows.
In the second section, we illustrate some background knowledge. From the third to the sixth section, we introduce typical estimation methods of correlation networks, conditional correlation networks, mixture networks, and differential networks accordingly. After that, we discuss the existing challenges to microbiome interaction network inference and aspects that can be further studied in the future. The detailed algorithm procedure of each method is summarized in Supplementary Appendix SA1.
2. Background
Sequencing depth is defined as the total sequencing count summed over all taxa of a sample and can vary across samples. Such variation of sequencing depth may be mainly caused by experimental techniques. Considering this fact, the microbial count data only carry information of relative abundances instead of absolute abundances of species, that is, the compositionality of microbiome data. First denote the observed count matrix as 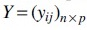 and the unobserved absolute abundance matrix as
and the unobserved absolute abundance matrix as  . Let
. Let
then the relative abundance matrix is  . The sample version of
. The sample version of

Thus, the correlation is characterized by a negative trend, even though the true abundances may be independent. Such problems can be managed by ratio transformation such as the centered log-ratio (clr) transformations and the additive log-ratio (alr) transformations.
Moreover, the number of features in the microbiome data is usually far larger than the sample size. In other words, microbiome data are high dimensional. This high dimensionality can lead to a heavy computational burden. Algorithms need to be designed carefully for efficient computation. Since the sequencing technique keeps improving, the scalability onto high-dimensional data having a larger scale is necessary. However, it is interesting to notice that from another perspective, high dimensionality plays a positive role in the analysis of compositional data. Although the number of features increases, the influence of false negative correlation trends may alleviate, which is called as “the blessing of dimensionality.”
Another feature of microbiome data is excessive zeros. In fact, zeros in the count data can be generally divided into two types. One type results from taxa that are present, but in proportions too low to be detected, and such zeros are often called technical zeros. Another type caused by the true absence of certain taxa, leading to essential zeros, is also called structural zeros (Peschel et al., 2020). Some methods choose to model these zeros, but the performance depends on the rationality of distribution assumptions.
In addition, many methods add a pseudo-count to avoid invalid computation. However, the choice of pseudo-count is subjective and may affect the performance of the method. Moreover, some methods choose to filter some nonsense taxa to preserve more informative features to handle problems caused by excessive zeros.
The composition of microbial communities can drastically vary from hosts to environments (Jiang et al., 2019). Thus, when collecting the data of many samples, a taxon that is relatively abundant in one sample may not be observed in other samples. This is the sparsity mentioned earlier. Another phenomenon is the tremendous variation among the dominant taxa collected in just one sample. This is termed as taxonomic heterogeneity (Jiang et al., 2019). In addition to taxonomic heterogeneity, multiple underlying microbial networks may determine the sample-taxa matrix in reality, rather than one single network. For example, in the human gut, the interaction between two taxa may depend on the enterotypic context of the individual's gut microbiome that can vary from individual to individual (Jiang et al., 2019). This is another type of heterogeneity that is functional.
3. Correlation Network
To identify the interactions among members of natural microbial communities, correlation analysis is proposed to measure the dependencies among microbes. However, the observed data only provide relative fractions of species. For compositional data, standard correlation analytic methods could result in spurious correlations. Therefore, various specialized methods based on compositional data have been developed to analyze the microbiome correlation network. See Table 1 for summary of correlation network methods.
Summary of Correlation Network Methods
3.1. SparCC
3.1.1. High-level idea
SparCC (Friedman and Alm, 2012) is just about the earliest method that takes compositionality into consideration. It introduces correlation between log-transformed absolute abundances based on compositional data. Then based on assumptions that the number of different OTUs is large and the actual correlation network is sparse, SparCC estimates the linear Pearson correlations between the log-transformed components via approximation. It proposes both a basic procedure and an iterative version to infer such linear Pearson correlations. The iterative one identifies component pairs with high correlations at each step to guarantee sparsity until the identification is stable.
Comment. SparCC estimates the correlations of log-transformed absolute abundances without the involvement of relative information, but there are several limitations of this method. First, although absolute abundances cannot be observed, SparCC can be free of compositional bias using this concept, implying that it relies heavily on the quality of component counts data. Second, the inferred covariance matrix by SparCC cannot be guaranteed positive definite, and correlation coefficients may not locate in
3.2. CCLasso
3.2.1. High-level idea
Similar to SparCC, CCLasso (Fang et al., 2015) considers the compositional property of the observed data in correlation analysis explicitly, and it has an extra advantage in that the estimated correlation matrix of the latent variables for compositional data is positive definite. CCLasso estimates correlations of log-transformed absolute abundance via weighted least-square regression with
Comment. However, the limitations of CCLasso are similar to those of SparCC. It also needs reliable component fraction estimation, only explains linear relationships, and does not offer theoretical performance guarantees. In addition, in terms of managing excessive zeros, CCLasso also adopts a simple pseudo count to avoid zero components, which may affect its performance. Lastly, the performance of CCLasso may not be satisfying if the number of taxa is much larger than that of samples.
3.3. REBACCA
3.3.1. High-level idea
REBACCA (Ban et al., 2015) identifies important co-occurrence patterns by finding sparse solutions. Different from SparCC, REBACCA constructs a linear system that is equivalent to log-ratio transformations and solves the system by using the
Comment. Similar to SparCC, REBACCA estimates pairwise relationships of log-transformed absolute abundances, so it is free of compositional bias. However, the iterative process may not scale to p, resulting in a high computational requirement. Besides, no theoretical performance guarantees are provided for REBACCA to work effectively. In addition, REBACCA may also fail when the sample size is small and it can be strongly affected by noise.
3.4. COAT
3.4.1. High-level idea
To solve the problems existing in the methods cited earlier and to provide statistical insights into the issue of identifiability and the impacts of dimensionality, COAT (Cao et al., 2019) was developed to estimate the sparse covariance matrix in a principled way and does provide theoretical guarantees on support recovery. It shows that the basis covariance is approximately identifiable if it belongs to the large sparse covariance family. Under the assumption that the basis covariance matrix is sparse, a composition-adjusted thresholding estimator is defined.
Comment. COAT is equivalent to thresholding the sample clr covariance matrix. So, it is free of optimization and efficient even when p is large. This work also fills the gap of large covariance estimation on constrained data by adopting covariance thresholding methods to compositional data. The work of COAT includes explicit statistical assumptions that are needed for it to be efficient, and it guarantees its performance theoretically. However, the estimated correlation relationship by COAT is linear. And there is no direct way for COAT to handle excessive zeros.
4. Conditional Correlation Network
Taking the compositionality of microbiome data into consideration, a microbial interaction network describes the pairwise correlations between two taxa. To restate, such correlations can be divided into direct interactions and indirect interactions. Indirect interactions involve two taxa correlated by some confounding factor, such as a certain environment condition, or a third species. Direct interaction means the nonexistence of confounding factors, and one taxon influences another directly. Since direct interactions are usually more fundamental and intrinsic (Friedman, 2004), methods modeling the conditional dependencies among taxa given all other taxa have gained much attention.
To infer such conditional dependencies, many methods turn to estimating a probabilistic graphical model in which each node represents a taxon, and each edge is undirected, indicating the conditional dependency of two taxa (Chen et al., 2020). If data follow a distribution in the exponential family, a straightforward statistical interpretation of such models can be made (Wainwright and Jordan, 2008). Taking the multivariate Gaussian distribution as an example, the element of the inverse covariance matrix is zero if and only if its two corresponding variables are conditionally independent given all other variables.
Therefore, to infer the conditional correlation network where an edge between two nodes represents conditional dependency given all other nodes, many researchers turn to estimating the nonzero entries of the inverse covariance matrix. The inverse covariance matrix is also termed as precision matrix. For real microbiome data, which are high-dimensional and sparse, the calculated covariance matrix is usually degenerate. In addition to this, we can only utilize the observed compositional data, which also makes estimating the precision matrix under absolute abundances challenging. See Table 2 for summary of conditional correlation network methods.
Summary of Conditional Correlation Network Methods
4.1. SPIEC-EASI
4.1.1. High-level idea
SPIEC-EASI (Kurtz et al., 2015) estimates the covariance of the log-ratio transformed absolute abundance approximately by calculating that of clr transformed data. It focuses on direct interactions and estimates’ conditional independence by inferring an underlying graphical model. The overall inference procedure of SPIEC-EASI includes estimating graphical models, as well as selecting neighborhoods and sparse inverse covariance. It is worth noting that SPIEC-EASI scales as the dimension of data grows and can recover the graph even when
Comment. The inference engine of SPIEC-EASI has the advantage of incorporating prior knowledge about the underlying data or network structure from independent scientific studies in a principled manner. Nevertheless, this method has several weaknesses. First, SPIEC-EASI relies heavily on the conditional number of the inverse covariance matrix. It also ignores the uncertainty of estimates, which assumes that the proportion of a taxon in a sample approximates the true value. Second, it is difficult for clr transformation to handle excessive zeros, and the transformed data are not even close to obeying Gaussian or sub-Gaussian graphical models. Third, the consistency of estimated covariance matrix cannot be guaranteed.
4.2. gCoda
4.2.1. High-level idea
gCoda (Fang et al., 2017) assumes that the absolute abundances follow a multivariate log-normal distribution and that the underlying network is sparse. Therefore, it can model the generation of relative abundance by a logistic normal distribution, and then estimate conditional dependence structures, which indicate direct interactions in microbial communities by maximum likelihood, using
Comment. gCoda is just about the first method to derive the likelihood of compositional data for Gaussian graphical models, However, one shortcoming of gCoda is that the likelihood function is nonconvex. Besides, the consistency of the estimator cannot be guaranteed.
4.3. MPLasso
4.3.1. High-level idea
MPLasso (Lo and Marculescu, 2017) uses biological knowledge mined from the literature as prior information in the graphical Lasso. The prior information including microbial co-occurrence patterns and associations is searched directly from a large amount of scientific literature by using automated text mining. It is utilized to restrict the search space to get more possible existing associations and make interaction inference more reliable.
Comment. MPLasso proposes to use experimentally verified biological knowledge as prior knowledge via text mining to enhance accuracy in inferring associations. Prior information plays a role in penalizing the elements of precision matrix, which may not dominate the results but restrict the search space. If the prior information mined is rare, then the method may be considered a graphical lasso problem. However, it has not been tested on a dynamic model of microbial communities. Extending MPLasso to estimate associations among communities may, therefore, require a new algorithm.
4.4. CD-trace
4.4.1. High-level idea
Considering that graphical lasso is not computationally efficient, D-trace loss (Zhang and Zou, 2014) is, hence, proposed. It is a convex and smooth loss function with a unique minimizer at the inverse covariance matrix. The sparse precision matrix can then be estimated by minimizing the lasso penalized D-trace loss under a positive-definite constraint. D-trace loss can be viewed as an analogue of the least-squares loss used in regression to estimate the precision matrix. Based on D-trace loss, CD-trace (Yuan et al., 2019) introduces an empirical loss function for compositional data called compositional D-trace loss. Under the positive-definite constraint, minimizing lasso-penalized CD-trace loss can get a sparse matrix estimator for the direct microbial interaction network.
Comment. The loss function proposed in CD-trace is convex, which makes the solution easier than that in gCoda. However, it has several limitations. First, during application, CD-trace adds a pseudo-count to avoid zero counts, which may result in inflated composition and the Gaussian distribution assumption may be oversimplified. This method still needs further study in contending with excessive zeros and information from these zeros. Second, theoretical support for the consistency of the estimator is absent. Third, the computational complexity of CD-trace is
4.5. CDTr
4.5.1. High-level idea
CDTr (He and Deng, 2019) is another loss function based on D-trace loss. Similar to CD-trace, CDTr also assumes that the log-transformed absolute abundance follows multivariate normal distribution. The sparse matrix estimators are defined as the minimizer of the corresponding lasso penalized loss, which has a unique solution under the exchangeable condition. For numerical solution, CDTr is based on the Alternating Direction Method of Multipliers (ADMM) algorithm to obtain a straightforward application.
Comment. We should consider the following limitations of CDTr. First, CDTr has a more concise form than CD-trace loss, but it still cannot guarantee the consistency of results. Second, the exchangeable condition used in CD-trace and CDTr needs theoretical verification and its biological reasonableness needs further checking.
4.6. MInt
4.6.1. High-level idea
MInt (Biswas et al., 2016) proposes a Poisson-multivariate normal hierarchical model to capture the conditional correlation structure. Confounding factors are controlled at the Poisson layer, and the multivariate normal layer with an
Comment. MInt is based on a Poisson-multivariate normal model, which models the Poisson mean as a multivariate normal random variable, leading to the flexible mean-variance structure. It can also readily handle over-dispersion. When inferring direct interactions, it takes controlling for covariates into consideration. However, these potential confounding variables must be diligently measured; otherwise, indirect interactions may be regarded as direct interactions. Besides, MInt directly handles count data without considering the compositionality of microbiome data explicitly.
4.7. mLDM
4.7.1. High-level idea
mLDM (Yang et al., 2017) is a metagenomic Lognormal-Dirichlet-Multinomial hierarchical model. It first models the count matrix with a multinomial distribution and then uses a Dirichlet distribution to model the multinomial probabilities. Lastly, a multivariate log-normal distribution is utilized to fit the absolute abundances of samples. mLDM estimates both the conditional associations among microbes and the associations between microbes and environmental factors and considers compositional bias and variance of the metagenomic data. The sparse association network is estimated by sparse learning techniques.
Comment. We should take the following limitations of mLDM into consideration. First, mLDM assumes that microbes have linear relationships with environmental factors, which may not actually be the case. Second, many parameters are introduced in mLDM, which can limit its efficiency and scalability. The complexity of mLDM also reduces its interpretability. Third, mLDM lacks theoretical justification of the estimators.
4.8. HARMONIES
4.8.1. High-level idea
Considering that microbiome sequencing data usually have different sequence depths across samples, many zeros, and over-dispersion, normalization is an important step before analysis. Unlike traditional normalization, which gets the compositional data first and then applies log-ratio transformation (Kurtz et al., 2015), HARMONIES (Jiang et al., 2020b) is a model-based normalization method designed for microbiome count data. It uses the zero-inflated negative binomial (ZINB) model with the Dirichlet process as a prior (DPP) to normalize data. After normalization, HARMONIES estimates the partial correlations to infer direct associations among taxa.
Comment. HARMONIES takes into account excessive zeros and over-dispersion by modeling the count data with a ZINB distribution. It also takes sample heterogeneity into account by further implementing DPPs. However, in some cases, the sample size obtained is always small, and this limitation has a direct effect on the estimated normalized matrix from the ZINB model.
4.9. BC-GLASSO
4.9.1. High-level idea
To reduce bias in the estimator coming from the proportion-based approach, BC-GLASSO (Jiang et al., 2020) directly models compositional count data by a multinomial distribution. Then, it assumes that multinomial probabilities follow a logistic normal distribution. The procedure of BC-GLASSO is similar to that of SPIEC-EASI, but it defines the true covariance matrix based on multinomial probabilities after the alr transformation rather than abundances after clr transformation. Afterward, BC-GLASSO conducts a bias-correction procedure for the estimator of true covariance matrix, during which the unevenness of sequencing depths can be handled. Based on this bias-corrected estimator, the graphical lasso is applied to infer a sparse inverse covariance matrix.
Comment. The alr transformation used in BC-GLASSO has the advantage of leading to a positive definite covariance matrix and thus makes the precision matrix well-defined. BC-GLASSO is, in fact, a hierarchical model. The multinomial distribution can capture technical variation of the count data, and the logistic normal distribution can detect biological variation of microbiome composition. BC-GLASSO considers the bias induced by the estimator and corrects such bias. However, we should notice that it also has several drawbacks, First, BC-GLASSO does not now take covariates, such as sex and age, into consideration. Second, since alr transformation needs a reference taxon, the potential interactions between the reference taxon and other taxa may not be estimated and analyzed by BC-GLASSO.
4.10. CompGlasso and its reference invariant version
4.10.1. High-level idea
CompGlasso (Tian et al., 2020a) also adopts the logistic normal multinomial distribution to model the count data, which is two-level hierarchical. However, the objective function used in it is derived from log-likelihood, which is different from that of BC-GLASSO. Alr transformation involves choosing a reference taxon, which may affect network estimation. Therefore, further analyses are needed to determine whether using a different reference taxon could change the nature of the estimated network in compositional graphical lasso and further affect its robustness. The reference-invariance property established (Tian et al., 2020) shows that the specific submatrix of the precision matrix is invariant to the choice of reference taxon. According to this property, the reference-invariant version of compositional graphical lasso is proposed, which only penalizes the invariant submatrix (Tian et al., 2020).
Comment. CompGlasso establishes the theoretical convergence of the whole algorithm, but the theoretical property of the estimator remains to be elucidated. The reference-invariant property clearly shows how different reference taxon choices affect the estimated network. Based on this property, a reference-invariant optimization problem is designed to make the alr transformation free from the choice of the reference taxon. However, the empirical reference-invariant inverse covariance matrix and network may depend on the algorithm used to optimize the objective function.
5. Mixture Network
The methods described earlier all assume that the sample-taxa matrix is only related to a single network topology and set of edge weights. A sample-taxa matrix, however, is constructed by various samples in which taxa may be involved in more than one biological network. Moreover, different environmental conditions can lead to different association networks. In particular, for human gut microbiome data, other variables, such as age, sex, and diet, can also influence microbial network interactions, changing them according to these variables. See Table 3 for summary of mixture network methods.
Summary of Mixture Network Methods
clr, centered log-ratio; MCMC, Markov Chain Monte Carlo; MPLN, Multivariate Poisson Log-Normal.
5.1. MixMPLN and MixMCMC
5.1.2. High-level idea
MixMPLN is a mixture model that comprises K Multivariate Poisson Log-Normal (MPLN) distributions (Tavakoli and Yooseph, 2019a). The mixture model directly tackles the count data, and the precision matrix of each component represents a microbial network. The MixMPLN framework is also extended by adding
Comment. MixMPLN tackles the count data directly, which considers different microbial clusters. Technical covariates, or environmental factors, which may influence a microbe network, can be included in the MixMPLN structure to control for confounding factors. However, there are several drawbacks of these two methods. First, as a mixture model, MixMPLN contains many parameters that can increase the time of optimization. Second, although MixMPLN could identify both positive and negative interactions between pairs of taxa for selected threshold values, the biological significance of these interactions should be investigated further. Lastly, the MCMC process in MixMCMC may be time-consuming.
5.2. MixGGM
5.2.1. High-level idea
MixGGM applies a log-ratio transformation to the sample count matrix and models the data as a mixture of Multivariate Gaussian distributions (Tavakoli and Yooseph, 2019). The iterative process of estimating parameters is similar to that in MixMPLN. The authors also extend MixGGM by adding sparse assumption onto the precision matrix for each multivariate Gaussian distribution. This is realized by applying graphical lasso to all K components.
Comment. MixGGM assumes that the covariance matrix of clr transformed data approximates that of log-transformed absolute abundances. Therefore, it may share the same problem as SPIEC-EASI, since approximation strongly depends on the conditional number of the inverse covariance matrix. Besides, the clr transformed data may not fit sub-Gaussian or Gaussian distribution well. Lastly, the computation complexity of MixGGM has not been well analyzed.
5.3. k-Lognormal-Dirichlet-multinomial model
5.3.1. High-level idea
As mentioned earlier, mLDM estimates both direct associations between two taxa and associations between taxa and environmental factors (Yang et al., 2017). However, this model only assumes one underlying biological network. Thus, the authors extended mLDM into k-Lognormal-Dirichlet-Multinomial model (kLDM) (Yang et al., 2020), which is a hierarchical Bayesian model and estimates multiple association networks with respect to specific environmental conditions. kLDM assumes that environmental factors regulate associations among microbes. These environmental factors tend to be stable given identical environmental conditions, but they vary with environmental conditions.
It also approximates the association pattern with multiple clusters, where each cluster represents an environmental condition. kLDM further assumes that the association networks are sparse. Then, it proposes a split-and-merge algorithm to estimate the number of environmental conditions and association networks.
Comment. For modeling, kLDM also takes compositional bias and the large variance of count data into consideration. Similar to mLDM, kLDM estimates both associations among microbes and associations between microbes and environmental factors. It can determine the number of environmental conditions automatically. However, kLDM has several limitations. First, the mathematical score used to merge clusters may not be sensitive enough to separate two clusters that represent two environmental conditions. Second, kLDM assumes that environmental factors follow a Gaussian distribution, which may not hold for categorical data. Lastly, kLDM shares a problem in common with mLDM in that too many parameters may lead to inefficient computation. This, in turn, results in unsatisfactory scalability of kLDM.
6. Differential Network
Mixture network inference methods estimate multiple underlying microbial networks simultaneously. In addition to considering the general case that multiple networks are associated with the sample-taxa matrix, discovering how the interaction network varies with environmental conditions or other confounding factors is another study that is of much significance to researchers. The differential network is often defined as the difference between two precision matrices. See Table 4 for summary of differential network methods.
Summary of Differential Network Methods
However, using the difference between these two estimators of a precision matrix as the estimated differential network can be problematic. One reason is that the estimator of a single precision matrix is usually obtained under the sparsity assumption. Directly minusing such two estimators may lead to false positive and negative interactions (Yuan et al., 2017). Even if the sparsity assumption were to be held, these methods would have most power in exploring strong interactions in a single condition. Thus, when detecting interactions that are not strong in one condition, but change strongly in other conditions, the power of these methods is limited (Yuan et al., 2017).
6.1. DCDTr
6.1.1. High-level idea
As mentioned earlier, CDTr incorporates the clr transformation into D-trace (Zhang and Zou, 2014) to derive the conditional correlation network. Further, to infer microbiome differential networks, while avoiding the use of unobserved absolute abundance data, DCDTr loss (He and Deng, 2019) is proposed by combining clr transformation with new D-trace (Yuan et al., 2017). DCDTr assumes that the difference between the precision matrices is sparse, rather than both matrices are sparse. The DCDTr loss proposed for differential network estimation is smooth and convex under the sparse differential network assumption. It can estimate the precision matrix difference directly with compositional data instead of estimating the precision matrices individually.
Comment. Similar to CDTr, the loss function proposed in DCDTr has a unique minimizer as the precision matrix if the exchangeable condition is satisfied. However, the reasonableness of the exchangeable condition still needs to be further studied. Most importantly, no theoretical guarantee on the consistency of the estimator is provided.
6.2. MDiNE
6.2.1. High-level idea
MDiNE (Mcgregor et al., 2020) is a Bayesian hierarchical model, which can estimate the co-occurrence network of two groups simultaneously with respect to a binary covariate and thus estimate the difference between them. MDiNE models the counts of individual taxa through a multinomial distribution, with its probabilities depending on a latent Gaussian random variable. In addition, the co-occurrence network among taxa is determined by a sparse precision matrix over all the latent items. The model is estimated through the Hamiltonian Monte Carlo algorithm.
Comment. MDiNE makes interval estimations for both parameters of the network and the difference between networks available. However, it has several limitations. First, an underestimation of true associations may occur when the sample size is relatively small. Second, MDiNE has no zero-inflation parameter, so it may encounter problems in controlling data with excessive zeros. Third, MCMC sampling may take too much time and increase computational burden.
6.3. Differential Markov random field analysis
6.3.1. High-level idea
Since previous graphical models cannot assign statistical significance for estimated associations, a Markov random field model and a hypothesis testing framework are proposed (Cai et al., 2019) to detect microbial network structure and differences between networks. They first discretize each composition vector into a binary one. Then, conditional dependencies between components in the vector are modeled by a binary Markov random field. To test whether two precision matrices are identical, they conduct a global test first. If the null hypothesis is rejected, then the multiple tests of each entry are performed. The binary Markov random field accounts for the excessive zeros and unit-norm constraint in the compositional data.
Comment. This model does not require the assumption of data distribution, which, in turn, increases its robustness. It can be used to interpret the co-occurrence and co-exclusiveness of the microbial communities naturally. And it can assign statistical significance to each element of the differential network. Nevertheless, there are several drawbacks that may limit its usage. First, this method is strongly affected by the discretization threshold chosen. Taking different cut-off values to transform raw data into binary data may cause different results. Second, each element of the discretized vector can only take two values. It should be extended to study the case in which each node can take more than two discrete values. Lastly, the consistency of results needs to be further examined, such as results when dichotomizing the data in different ways.
7. Discussion
7.1. Ways to handle environmental confounding factors
Environmental factors, such as pH, humidity, temperature, and soil type, can strongly affect the composition of the microbial community. Understanding interactions among microbes and between microbes and environmental factors is of great importance. However, whether the edge in the microbial network is caused by an environmental factor or a direct interaction between two taxa is hard to distinguish, and many detected associations between microbes may be spurious without considering environmental factors (Yang et al., 2017).
Microbial network inference can handle environmental factors by regarding them as additional species and calculating their association with microbes, or treating them as extra variables in regression and inferring the association of the remaining abundances unaffected by the environment (Faust and Raes, 2012), such as mLDM (Yang et al., 2017) and kLDM (Yang et al., 2020). In addition, as mentioned in Faust (2021), we can cluster samples according to key environmental factors and construct networks for each sample group separately to decrease environmental variation within a group.
Multiple ways are available to predict the associations between species and environmental factors, but which one to use relies on concrete goals during the research. For instance, treating environmental factors as additional nodes helps us realize how they influence microbial community composition, whereas stratifying samples or regressing out environmental confounders is beneficial for inferring microbial interactions. However, the problem lies in establishing a reliable evaluation system of various treatments to properly manage environmental factors (Faust, 2021).
7.2. Ways to detect complex relationships
As mentioned earlier, methods that estimate interaction by means of Pearson correlations (Friedman and Alm, 2012), covariance matrix, or precision matrix all assume that the microbial correlation relationship is pairwise linear, which is not always the case. Sometimes, nonlinear complex interactions may occur in the microbial community. An interaction among some species could be influenced by an additional one (Faust, 2021), and one species could be affected by other species (Faust and Raes, 2012).
These cases could not be handled correctly with direct pairwise interaction detection methods mentioned in this article. In fact, high-order interactions have been shown to affect the microbial community both in simulations (Bairey et al., 2016) and in experiments (Gould et al., 2018). Hence, it is necessary to find ways to manage complex relationships, which could provide evidence for nonrandom co-association patterns (Cardona et al., 2016).
We can detect complex relationships by only considering simple relationships first. After the model is constructed, the biases between model estimations and actual microbial community behaviors may indicate the presence of complex relationships (Faust, 2021). However, improper model construction also can lead to the skewing of model predictions (Billick and Case, 1994), which makes it hard to distinguish the existence of complex relationships.
We can also handle complex relationship inferences by association rule mining, which involves enumerating all logical rules in the presence or absence of data to find valuable rules (Faust and Raes, 2012). For instance, if species A exists only in the presence of both species B and C, then neither the interaction between A and B nor the interaction between A and C exists unless a third species appears. This can be regarded as an extreme example of relationship correction (Faust, 2021).
People have reported several association rules containing more than two taxa in the past. However, possible reasons behind relationships detected may include true complex relationships and confounding results such as the assembly of pairwise interactions and overfitting (Faust, 2021). Thus, it is still a problem to uncover the underlying reasons that can lead to the detection of complex relationships, while still avoiding meaningless results.
Another approach worth trying is multiple regression, which predicts the abundance of a species through incorporating the abundance of other species (Faust and Raes, 2012). The principle of multiple regression is simple and clear and is, therefore, widely applied. However, this method is limited by the difficulty of discovering the implication of results when we sort factors through automatic feature selection methods such as sparse restriction. In this way, although we may be able to provide a roughly accurate prediction of the abundance of the species mathematically (Faust and Raes, 2012), the existence of causality in biology is doubtful.
In addition, people also attempt to capture complex forms of ecological interactions through the mutual information criterion, which measures the additional information known about one variable when the other variable is given (Butte and Kohane, 2000). Maximum information coefficient (MIC) is widely used to capture novel correlations on large datasets (Cardona et al., 2016), which puts mutual information into a continuous distribution and identifies diverse interactions between variables, both linear and nonlinear (Reshef et al., 2011).
However, sample size must be large enough to calculate the value of MIC accurately, whereas this can increase the difficulty of implementation (Li et al., 2016). Besides, recent research has questioned the equitability and statistical performance of MIC (Gorfine et al., 2012).
Finally, a microbial network involves complex relationships that likely consist of edges jointing more than two nodes. Consequently, it is not an easy task to visualize them (Faust and Raes, 2012). Hypergraphs should be taken into consideration, and how to investigate and explain them needs further exploration (Faust, 2021).
7.3. Tradeoff between model accuracy and computational complexity
We realize the fact that simple models may fail to fit the microbial community well, whereas complex models may lead to computational burden. For instance, compared with Pearson correlation, SparCC is proven to be more suitable for avoiding false interactions, while inducing higher computational complexity (Matchado et al., 2021). Further, correlation-based methods usually cannot distinguish direct from indirect interactions. To solve this problem, a large number of methods based on conditional dependence have been developed.
Compared with correlation-based methods, some conditional dependence methods have a sharp increase in computational complexity, which results in unsustainable running times (Matchado et al., 2021), such as hierarchical Bayesian models, for instance, mLDM (Yang et al., 2017) and kLDM (Yang et al., 2020). Therefore, the tradeoff between model accuracy and computational complexity is another point we should take into account.
To solve this dilemma, we should consider the computational burden brought by concrete datasets and available computation resources when choosing a proper analytic method to use. We should also consider whether it is feasible for the microbiome to be analyzed on a higher taxonomic level.
Since different methods can detect different edges in the same data (Weiss et al., 2016), it is assumed that model accuracy can be improved by using a combination of various analytical tools. As a matter of fact, some studies have reported experiments on linearly modeled data with foregone engineered interactions (Weiss et al., 2016). Compared with most individual methods, it has been shown that the accuracy of the ensemble method is significantly improved at the cost of sensitivity. Hence, when the given research requires us to obtain results of high precision, for instance, utilizing hypotheses of microbial interactions to identify co-culturing, integrated methods should be applied.
7.4. Lack of universal standardized simulation and evaluation
Simulated datasets are often needed to test the performance of methods. Various ways are currently available for simulation. One famous approach is generating data from statistical distributions, such as the logistic normal distribution adopted by CCLasso (Fang et al., 2015), COAT Cao et al. (2019), and gCoda (Fang et al., 2017). Another widely used method involves adopting specific models, especially differential equations, such as the famous generalized Lotka–Volterra model (Buffie et al., 2015; Bucci et al., 2016; Venturelli et al., 2018).
However, we are still confounded by the lack of a universal standard for simulating microbial community with ground-truth correlations. This makes it difficult to evaluate microbial network inference methods in a unified way since the performance of the proposed methods is largely affected by the data generation approach. If the artificial data are generated in a way that approximates assumptions of the microbial correlation network inference model, then the model would gain a relatively good performance. That is, one way of simulating data would favor methods similar to its generation approach (Faust, 2021). However, the true generation process of microbial data in reality is unknown, which may be more complex than current simulation approaches.
To avoid the evaluation bias caused by a single simulation method and judge the performance of various methods more objectively, it is necessary to adopt different simulation methods when testing. It is also important to separate developing new microbiome correlation network inference methods from comparison studies to ensure neutrality (Faust, 2021). Weiss et al. (2016) compare the performance of eight correlation techniques, as well as their ability of handling the issues in compositional data. Hirano and Takemoto (2019) investigate more recent methods, such as REBACCA (Ban et al., 2015) and CCLasso (Fang et al., 2015).
Comparative studies show that correlation estimation methods based on compositional data do not always outperform classical methods, such as Spearman correlation-based and MIC-based methods. This may result from the fact that newly proposed methods are developed under certain conditions or for particular problems.
7.5. Lack of benchmark microbial network dataset
In addition to simulated datasets, real datasets with verified biological microbial associations are also vital to evaluate those interaction inference methods. Biological gold standard network datasets for interaction inference are considered those datasets that have available sequencing data and validated known interactions. Lima-Mendez et al. (2015) built a literature-curated collection of 574 interactions in marine eukaryotic plankton, in which both mutualism and parasitism interactions are included. Besides, Durán et al. (2018) examined root-associated microbial communities and detected some microbial interactions that have been verified experimentally. However, benchmark datasets for microbiome interaction network inference are still scarce.
Several obstacles interfere with constructing benchmark real datasets. First, some interactions that happen in the real world may not be observed at all. Thus, the verified interactions are inadequate and incomplete. Using these datasets as benchmarks to test microbiome network inference methods would improve our evaluation of how these methods discover known associations. Even if a method could catch sight of underlying interactions among various taxa, such interactions may be viewed as false positives since they were not observed experimentally.
Second, as previously pointed out, the interactions can be either direct or indirect. In reality, they are complex, and it is difficult for some methods to tell which kind of interactions are inferred. Especially, correlation network inference methods can obtain direct interactions or indirect ones, such as high-order interactions where associations among some species are altered by an additional species. The gap between inferred interactions and expected ones brings a challenge to the development of benchmark microbial datasets.
8. Conclusion
Microbial interaction estimation provides an insight into the complex interplay that exists in microbial communities and the connections between the microbiome and its living environment. Statistical computational methods can infer such association networks and guide further biological studies.
In this review, we follow the development of microbiome network inference and analyze typical methods of correlation networks, conditional correlation networks, mixture networks, and differential networks. These methods tackle inference problems from various aspects, and they are based on different assumptions, even while facing some common challenges coming from the compositional data.
In practice, we recommend the following principle to choose a proper method in a certain scenario. First, taking factors such as type of interactions, heterogeneity of data, and the dynamics of networks into consideration, we should decide which network structure we are concerned about. Second, we should consider the characteristics of data, such as data distribution, the existence of noise, as well as the availability of prior information. Finally, sample size and computation complexity are also determining factors for users to make a decision.
After estimating correlations among taxa, their biological interpretation is further required, which is not an easy task. Most current methods fail to recognize the nature of relationships between microbes and cannot credibly distinguish authentic biological correlations from artifact ones. More specific explanations require the support of biological experiments and the literature.
In addition, we have mentioned that excessive zeros as well as the curse of dimensionality bring in challenges in microbiome compositional data network inference. To mitigate their negative effects, people often choose to group relative abundances on a higher taxonomic level. However, this may lead to hiding insights into smaller communities.
In addition to types of networks analyzed in the main paper, microbiome network inference methods can be improved by accounting for more factors affecting the living microbiome, estimating more complex relationships, and considering time-variable networks. A more advanced network theory is required to offer a further comprehensive understanding into the mechanism for the microbiome to influence the host and vice versa.
For instance, we should not ignore the fact that microbes from various life domains may have mutual effects and usually live in the same habitat. Compared with a single domain, joint cross-domain network inference can result in marked differences of the features and integral structure of the network. Thus, it is a vital developing direction to extend existing methods to cross-domain network analysis.
Nowadays, microbiome multi-omics datasets can be built by collecting multiple types of biological data, such as metagenomics, metatranscriptomics, 16S, mass spectrometry-based metaproteomics, and metabolomics (Helbling et al., 2012). Different types of biological data provide information from different views, so consolidating them together can bring a more comprehensive view. However, in reality, it is rare that all samples have all types of features measured, owing to the difficulty of gathering multi-omics data (Jiang et al., 2019). Therefore, integration methods should be able to manage data with missing features and not rely on paired multi-omics data.
How to make methods more scalable with less computational complexity, while, at the same time, guaranteeing efficiency, is also a task for future studies. As discussed earlier, some challenges in benchmarking real microbiome datasets need resolution to simulate reliable data for evaluation and propose universal valuation standards. All these problems require improvements in biological techniques and statistical methods together.
Footnotes
Acknowledgments
The authors sincerely thank all the reviewers and editors for their efforts and suggestions. They wish Professor Michael Waterman “A happy birthday!”
Author Disclosure Statement
The authors declare they have no conflicting financial interests.
Funding Information
This work was supported by the National Key Research and Development Program of China (2021YFF1200902) and the National Natural Science Foundation of China (12126305, 31871342).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.