
Abstract
We propose GRNUlar, a novel deep learning framework for supervised learning of gene regulatory networks (GRNs) from single-cell RNA-Sequencing (scRNA-Seq) data. Our framework incorporates two intertwined models. First, we leverage the expressive ability of neural networks to capture complex dependencies between transcription factors and the corresponding genes they regulate, by developing a multitask learning framework. Second, to capture sparsity of GRNs observed in the real world, we design an unrolled algorithm technique for our framework. Our deep architecture requires supervision for training, for which we repurpose existing synthetic data simulators that generate scRNA-Seq data guided by an underlying GRN. Experimental results demonstrate that GRNUlar outperforms state-of-the-art methods on both synthetic and real data sets. Our study also demonstrates the novel and successful use of expression data simulators for supervised learning of GRN inference.
1. INTRODUCTION
Inferring gene regulatory networks (GRNs) from microarray or single-cell RNA-Sequencing (RNA-Seq) gene expression data sets has been an active area of research for more than two decades. This topic is receiving renewed attention in the context of single-cell transcriptomic data (Chen and Mar, 2018; Kiselev et al., 2019; Pratapa et al., 2020). In contrast to bulk transcriptome data of prior years, scRNA-Seq data provide cellular level activity, although with higher levels of noise and more data sparsity (Vallejos et al., 2017).
Several GRN reconstruction methods that were originally developed for bulk transcriptional data have been applied (Irrthum et al., 2010; Huynh-Thu and Sanguinetti, 2015; Kim, 2015; Moerman et al., 2019) or adapted (Chen et al., 2015; Lim et al., 2016; Hamey et al., 2017; Woodhouse et al., 2018) to single-cell data, and new methods have been developed specifically for it (Aibar et al., 2017; Chan et al., 2017; Matsumoto et al., 2017; Specht and Li, 2017; Papili Gao et al., 2018; Sanchez-Castillo et al., 2018). For a recent review on the performance and comparative evaluation of various GRN methods for single-cell transcriptomic data, see Pratapa et al. (2020).
An important class of methods developed for GRN inference is based on unsupervised learning. GRNBoost2 (Moerman et al., 2019) and GENIE3 (Vân Anh Huynh-Thu et al., 2010), among the top performing methods (Chen and Mar, 2018; Pratapa et al., 2020), operate by fitting regression functions between the expression values of the transcription factors (TFs) and other genes. An alternative approach is to pose GRN inference as a graphical lasso problem with l1 regularization (Friedman et al., 2008).
All these approaches are primarily unsupervised in nature. Recently, simulators that generate synthetic scRNA-Seq data guided by GRNs have progressed significantly (Dibaeinia and Sinha, 2019; Pratapa et al., 2020). They can generate realistic data by modeling sources of variation in scRNA-Seq data such as noise intrinsic to the process of transcription, extrinsic variation indicative of different cell states, technical variation, and measurement noise and bias. These simulators have been primarily developed and applied to systematically benchmark GRN inference methods.
In this study, we propose to leverage GRN-guided simulators in a novel way to enable supervised learning of GRNs from scRNA-Seq data. Motivated by the recent successes in supervised neural network (NN)-based algorithms in learning graphical models (Belilovsky et al., 2017; Shrivastava et al., 2020), we propose a deep learning (DL) framework that takes expression data as input and outputs the corresponding GRN. For the purpose of supervised training of our framework, we use the SERGIO by Dibaeinia and Sinha (2019) simulator to generate a corpus of training examples containing gene expression data sets and the corresponding GRNs.
Our DL framework consists of two novel modeling choices. First, we leverage the expressive ability of NNs to capture the dependencies between TFs and the corresponding genes they regulate, by aptly using an NN in a multitask learning framework. Second, to capture sparsity of the GRNs observed in the real world, we design an unrolled algorithm for our framework. Unrolled algorithm is an emerging paradigm in machine learning that is gaining prominence in discovering sparse graphical models. Key advantages include (1) fewer parameters to be learned, (2) less supervised data points required for training, (3) comparable or better performance than state-of-the-art methods, and (4) more interpretability.
Unrolled algorithms have been successfully designed in recent studies, for example, RNA secondary structure prediction method E2EFold by Chen et al. (2020) and sparse graph recovery technique GLAD by (Shrivastava et al., 2020). Both the multitask learning NN and sparsity-related parameters are optimized jointly in our DL framework using supervision.
Our unrolled DL model, termed GRNUlar (pronounced “granular”) for
2. PROBLEM SETTING AND CHALLENGES
We consider the input gene expression data to have D genes and M samples,
2.1. Existing approaches
The common approach followed by many state-of-the-art methods for GRN inference is based on fitting regression functions between the expression values of TFs and each of the genes. Usually, a sparsity constraint is also associated with the regression function to identify the top influencing TFs for every gene.
In general, the objective function used for GRN recovery in various methods is a variant of the equation given hereunder. For all
Equation (1) can be viewed as fitting a regression between the expression value of each gene as a function of the TFs and some random noise. The simplest model will be to assume that the function fg is linear. One of the widely used methods, TIGRESS by Haury et al. (2012), assumes a linear function of the following form for every gene:
GRNBoost2 (Moerman et al., 2019) further uses gradient boosting techniques over the GENIE3 architecture to do efficient GRN reconstruction. Supervised learning methods for recovering the GRN by inferring the gene–gene interactions for all the possible gene pairs have also been recently explored (Yuan and Bar-Joseph, 2019; Razaghi-Moghadam and Nikoloski, 2020). Our approach differs from them as we formulate our framework to jointly optimize for all the interactions.
2.2. Drawbacks
There are two major drawbacks in current approaches that optimize for Equation (1). The first is in choosing the function fg, which can be improved further to better capture nonlinear relations and make the method more robust to noise in data. The second is tuning the sparsity-related hyperparameter for the GRN that usually requires an additional post hoc scoring step. Such scoring process to obtain the desired sparsity of GRN is suboptimal. A better approach would be to jointly optimize the sparsity along with discovering the underlying GRN.
3. THE PROPOSED GRNUlar FRAMEWORK
To overcome the aforementioned drawbacks, we propose a DL framework with the following three key components:
Choice of fg: We model fg using NNs that are able to learn expressive class of highly nonlinear functions (Goodfellow et al., 2016). Instead of the traditional viewpoint of considering a NN as a black box, we view the NN itself as a multitask learning framework. The path connections between the input neurons and the output neurons of the NN can be easily interpreted as the underlying GRN, where the multiple tasks correspond to the inference of TFs for multiple genes. Use supervision: We develop a DL model that leverages simulators to generate training examples for supervised learning. The training data consist of multiple input gene expression data sets and the corresponding GRNs. We hypothesize that tuning GRN recovery models under this supervision will lead us to better capture intricacies of real data, and potentially improve upon the unsupervised methods. Capture sparsity: We use the recently developed unrolled algorithm paradigm to design the deep architecture that can model the underlying sparsity as a parameter that can be learned under supervision.
3.1. NN modeling of regression functions
NNs are capable of representing rich classes of highly nonlinear functions. We combine the regression formulation in Equation (1) with NNs in a multitask learning framework (Ruder, 2017; Fig. 1) to learn multiple nonlinear regression functions estimating dependencies between each gene and the set of TFs. If there is a path in the NN from an input TF to the output gene, then the output gene is dependent on the corresponding TF. We thus want sparse NN weights
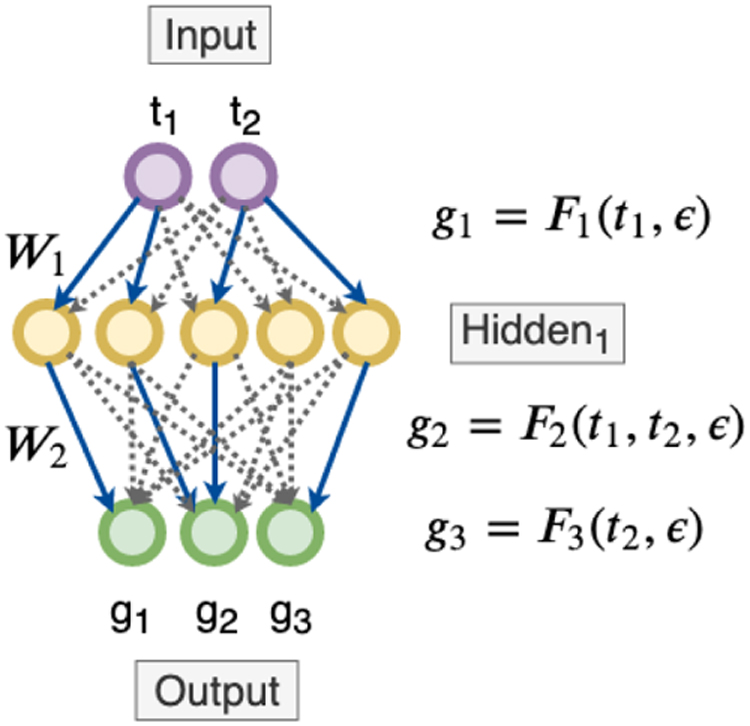
Using NNs as a multitask learning framework: We start with a fully connected NN indicating all genes are dependent on all the input TFs (dotted black lines). Assume that in the process of discovering the underlying sparse GRN, our algorithm zeroes out all the edge weights except the blue ones. Now, if there is a path from an input TF to an output gene, we then conclude that the output gene is dependent on the corresponding input TF. GRN, gene regulatory network; NN, neural network; TFs, transcription factors.
We can easily obtain the dependency matrix between input TFs and output genes as a matrix multiplication, where
This multitask NN architecture is superior to boosted decision tree-based formulations as it is more expressive, and does not need the additional post hoc scoring step. It is also more expressive than a simple nonlinear model with additive noise because the NN is jointly optimizing the regression for all the output genes (multitask learning) and this helps it capture the common dependencies between the TFs and output genes. This also makes the NN model more robust toward external noises as jointly optimizing for all the gene expression values will mitigate the effect of any expression value anomalies that seeped in during the experiments.
3.1.1. Designing the unrolled algorithm
Many NN representations can satisfy Equation (1), which leads to multiple possible GRNs. These GRNs will vary mostly in terms of the sparsity obtained and it is hard to tune for the desired sparsity of the GRN manually. Unrolled algorithms help resolve this problem as the sparsity-related hyperparameters (e.g., the weight of the l1 norm term) can be learned from supervision. Our aim is to get a deep model that optimizes the multitask learning NN along with learning the optimal sparsity pattern using the supervision provided from simulator data. We follow a similar procedure as the unrolled algorithm designed for sparse graph recovery by using the alternative minimization (AM) algorithm (Shrivastava et al., 2020).
3.1.1.1. Identifying the inductive bias
We wish to simultaneously fit the regression formulations in Equation (1) as well as learn the desired sparsity of the underlying GRN. One way to achieve this is to jointly optimize the regression error with an l1 penalty term over the dependency matrix. Thus, we consider the following nonlinear optimization objective function for the regression with l1 penalty

where
3.1.1.2. Using optimization algorithm as design template
We now identify the iterative updates of a suitable optimization algorithm. Since the mentioned objective is nonlinear, we will need an iterative approach to minimize it w.r.t.  . Now, alternatively minimize Z and
. Now, alternatively minimize Z and
The update of Z is of the form
3.1.1.3. Unrolling the iterations to get deep model
We now unroll the iterative updates to certain iterations, identify key learnable components, and treat the entire unrolled iterations as a single highly recurrent deep model. We identify the proximal operator

Visualizing the GRNUlar algorithm's architecture. It is a single deep model that is highly structured and recurrent. It takes gene expression data as input and outputs the corresponding GRN. GRNUlar,
Algorithm 1, GRNUlar-basic, provides a supervised learning framework for the unrolled model directly based on the updates of the AM algorithm, Equations (3) and (4). We typically require
We posit that, if we optimize for the first term of Equation (3) beforehand and obtain good initial values of
A note on backpropagation of gradients: While taking the
We typically require
The GRNUlar model can be thought of as having two stages, namely (refer Fig. 2 and Algorithm 2).
3.2. A note on parameterizing the NNs of unrolled algorithms
The general idea of NN-based parameterization: For the GRNUlar algorithm, we can further parameterize the optimizer update given in “fitDNN-fast” function of Algorithm 2 and learn it from the supervision provided, similar to
For instance, consider the example of parameterizing gradient descent optimizer that is realized using the
4. TRAINING THE GRNUlar FRAMEWORK
GRNs are typically sparse and so we want our loss function to be robust enough to recover sparse edges. There are many techniques developed to train in a cost-sensitive setting where in the data we have very few positive data points (edges present) versus large number of negative data points (edges absent) (Chawla et al., 2002; Thai-Nghe et al., 2010; Shrivastava et al., 2015; Bhattacharya et al., 2017). Since there are multiple metrics such as precision, recall, and F1 score that are commonly used for evaluation of the recovered GRNs, it will be useful to define a loss function that can find a desirable balance between them.
To address the mentioned concerns, we develop a differentiable version of the
where
A value of
We define a loss function between the predicted and true adjacency matrix as the combination of the mean square error (MSE) (or Frobenius norm) loss and the
The matrix
5. EXPERIMENTAL RESULTS
5.1. Methods compared and evaluation measures
We use the area under the receiver operating characteristics (AUROC) and the area under the precision recall curve (AUPRC) values for evaluation (Chen and Mar, 2018; Dibaeinia and Sinha, 2019) and comparison of various methods. We compared GRNUlar with GRNBoost2 (Moerman et al., 2019, GENIE3 (Vân Anh Huynh-Thu et al., 2010), and GLAD (Shrivastava et al., 2020).
GRNBoost2 and GENIE3 are representative of regression-based methods, and are among the top performers for single-cell expression data (Pratapa et al., 2020). We used the Arboreto package to run these algorithms (Moerman et al., 2019). We did extensive fine tuning of the hyperparameters for both the methods using the training/valid data and reported results on the test data. “Method+TF” indicates that TF information was utilized for GRN recovery.
GLAD by Shrivastava et al. (2020) is an unrolled algorithm designed for sparse graph recovery. It is based on unrolling the iterations of an alternate minimization algorithm for the graphical lasso problem. It fits a multivariate Gaussian distribution on the input gene expression data with an l1 normalization term. We modified the GLAD algorithm to take into account TF information, called GLAD+TF, by using a post hoc masking operation that only retains the edges having at least one node as a TF. We used the standard initialization as recommended by the authors.
We chose the number of unrolled iterations L = {15, 30}. For the GRNUlar model, we used the same initialization of the thresholding parameters
For both the unrolled methods, we chose two models based on AUPRC and AUROC results on the validation data. We use the scaled loss function [Eq. (7)] to jointly optimize for MSE and
5.2. Details of SERGIO simulator for clean and noisy settings
SERGIO provides a list of parameters to simulate cells from different types of biological processes and gene-expression levels with various amounts of intrinsic and technical noise. We simulated cells from multiple steady states. When simulating data with no technical noise (what we refer to as clean data), we set the following parameters: sampling-state = 15 (determines the number of steps of simulations for each steady state); noise-param
The parameters required to decide the master regulators' basal production cell rate for all cell types—low expression range of production cell rate
5.3. Evaluating GRN inference methods on synthetic data
We conducted an exploratory study to gauge the generalization ability of GRNUlar for the GRN inference task. To provide supervision, we used the SERGIO simulator. To create random directed graphs (GRNs), we first decided on the number of TFs or master regulators. Then, we randomly added edges between the TFs and the other genes based on sparsity requirements. Also, we randomly added some edges between the TFs themselves but excluded any self-regulation edges and maintained connectivity of the graph.
The graph is then provided as input to the SERGIO simulator to generate corresponding gene expression data. For the experiments in this subsection, we took train/valid/test =

Clean data setting of the SERGIO simulator with D = 100 genes. As the number of cell types increases from C = 2 to C = 10, we see that the AUPRC values increase in general. The unrolled algorithms in general outperform the traditional methods. AUPRC, area under the precision recall curve.
From the literature on sample complexity theory of sparse graph recovery (e.g., see, Ravikumar et al., 2011), we know that recovery of the underlying graph improves with increasing number of samples. Hence, we ran our experiments with varying number of the total single cells, M = {100, 500, 1K, 5K, 10K}.
We also observed that varying the number of cell types (corresponding to the number of clusters of the cells) of the SERGIO simulator considerably affects GRN inference results, so we also evaluated the methods by varying the number of cell types of the simulator C = {2, 5, 10}. We adjusted the number of cells per cell type to maintain the same total number of cells. Section 5.2 contains detailed description of SERGIO settings. For experiments in this subsection, each data point in the plots represents its value along with standard deviation over the test graphs.
5.3.1. Clean: simulated data with no technical noise
The “clean” gene expression data from SERGIO follow all the underlying kinetic equations but exclude all the external technical noises. These data can be considered as being recorded with no technical errors. Figure 3 compares different methods on their AUPRC performance on varying number of cells and number of cell types. For GRNUlar model we chose two-layer NN with p = 5, Hd = {40, 60, 100}, L = 15, and vary
5.3.2. Noisy: simulated data with technical noise
We evaluate on the more challenging and realistic noisy settings. We limit varying the technical noise to dropouts while keeping the default settings for other SERGIO parameters. For higher levels of dropouts, researchers sometimes resort to data imputation techniques (which attempt to recover the number of molecules being dropped) as a preprocessing step that marginally improves results. For these experiments, we report results without the imputation preprocessing step and compare all methods directly on the noisy data obtained from the simulator.
While training the models, we train on data with low dropout rates
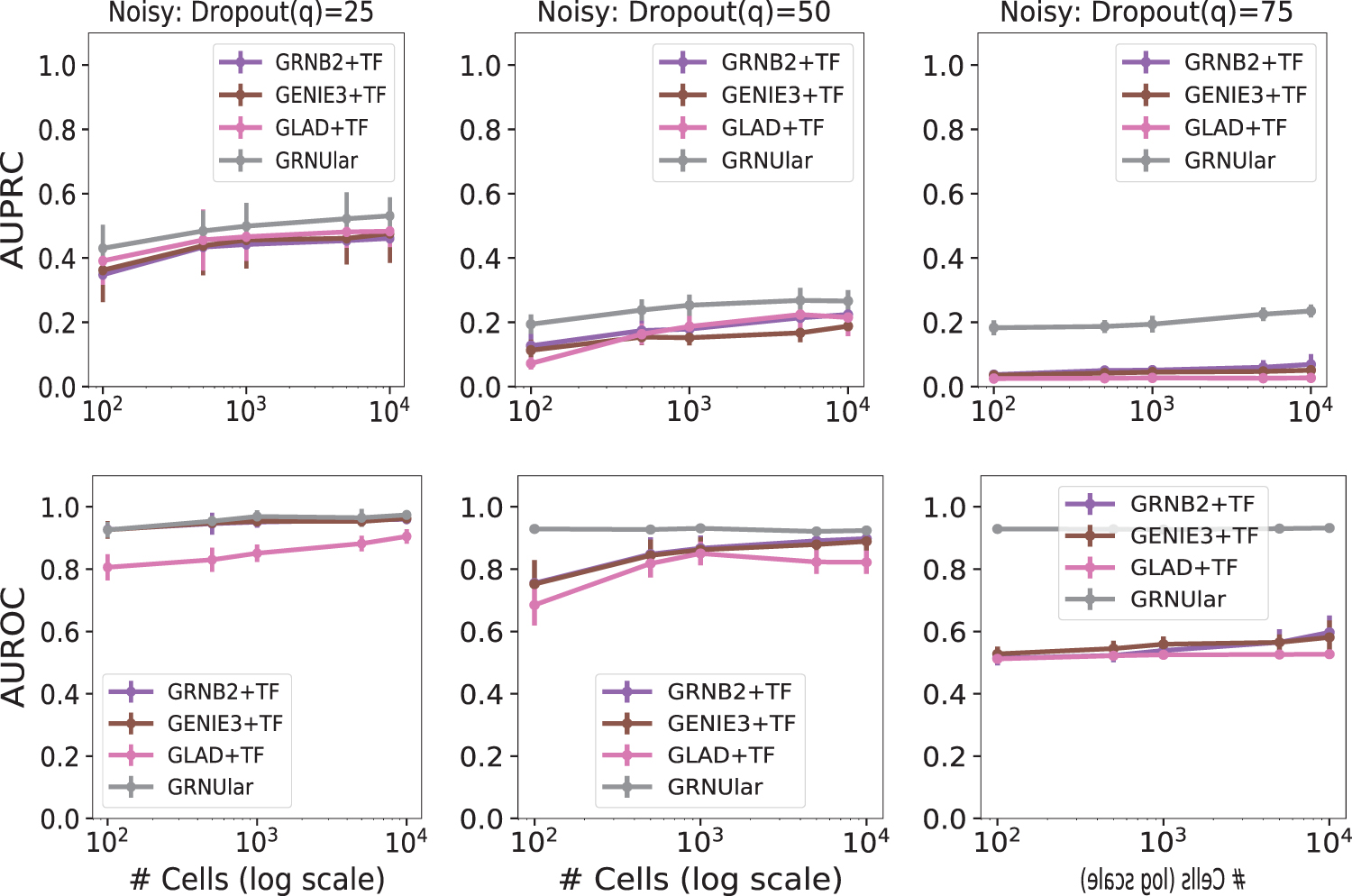
Noisy data setting of the SERGIO simulator with dropout shape = 20, D = 100, and C = 5. We vary the dropout percentile values as [25, 50, 75] in both the upper panels (AUPRC values) and the lower panel (AUROC values). Larger q corresponds to higher technical noise. GRNUlar has a clear advantage in noisy settings. AUROC, area under the receiver operating characteristics.
5.4. Realistic data from SERGIO: Escherichia coli and yeast
The challenge for data-driven models is to be able to generalize to real data sets. Thus, it is important to test the ability of the unrolled algorithms to generalize over different settings from that of the training. To perform this study, we make use of the realistic data sets provided by the SERGIO simulator. They provide three scRNA-Seq data sets DS1, DS2, and DS3 that are generated from input GRNs with 100, 400, and 1200 genes, respectively. These networks were sampled from real regulatory networks of Escherichia coli and Saccharomyces cerevisiae.
For each data set, the settings are number of cell types C = 9, total number of single cells M = 2700, and there are 300 cells per cell type. Each data set was synthesized in 15 replicates by re-executing SERGIO with identical parameters multiple times. The parameters were configured such that the statistical properties of these synthetic data set are comparable with the mouse brain, given in Zeisel et al. (2015).
We defined our training and testing settings such that there were considerable differences between them. We used all of the DS1, DS2, and DS3 data sets for testing. We train on data with settings similar to DS1, specifically the parameters such as production cell rates, decays, noise parameter, and interaction strength. We trained with no dropouts as opposed to 82% dropout percentile in the case of the DS data sets. The data sets DS2 and DS3 are completely different from training data (and DS1) in terms of the underlying GRN, as well as the corresponding SERGIO parameters are sampled from different range of values. For details, refer to table 1 and appendix tables S1, S3 in Dibaeinia and Sinha (2019) for more insight into the differences in SERGIO parameter settings.
Details of Expression Data from the BEELINE Framework
Total number of genes for each data is 500 (highest varying genes).
Experiments: hESCs, human embryonic stem cells; hHep, human mature hepatocytes; mDCs, mouse dendritic cells; mESCs, mouse embryonic stem cells; mHSCs-E, mouse hematopoietic stem cells-erythroid; mHSCs-GM, mouse hematopoietic stem cells-granulocyte-macrophage; mHSCs-L, mouse hematopoietic stem cells-lymphoid; TFs, transcription factors.
5.4.1. GRNUlar settings
We used a two-layer NN in the fitDNN-fast function for these experiments with a single hidden layer H1. Following our strategy to choose the dimensions,
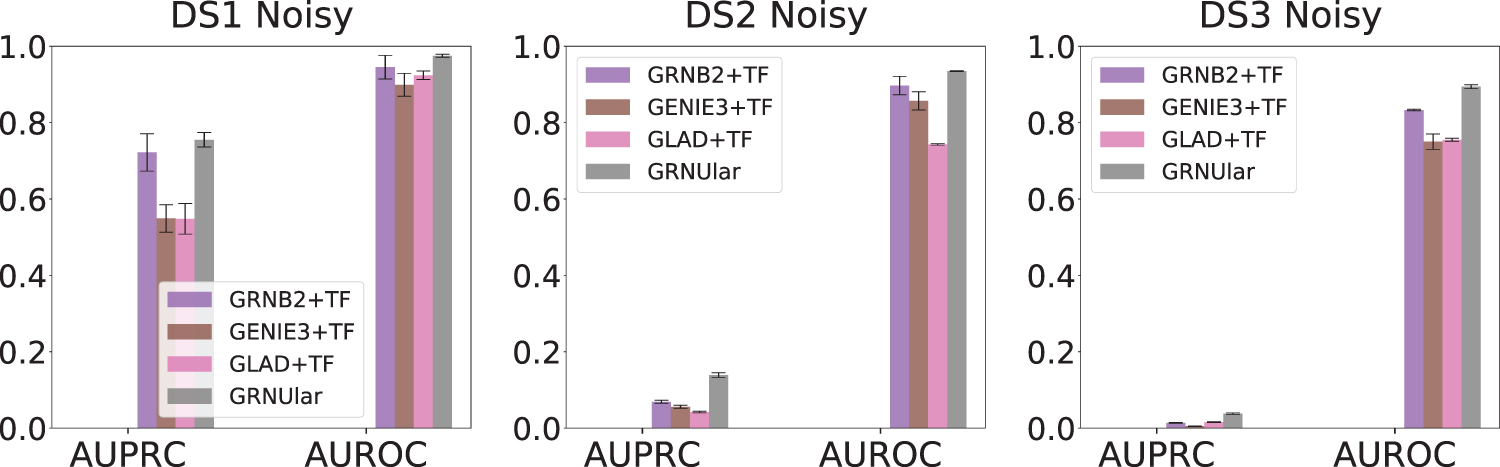
Realistic data from SERGIO of Escherichia coli and yeast—(noisy settings, dropout percentile = 82%). We report the average results over 15 test graphs in the noisy settings. GRNUlar gives notable AUPRC values and it outperforms other methods.
5.5. Real scRNA-Seq data sets
We evaluated 21 gene expression data sets from the human and mouse species and their corresponding ground truth networks (Pratapa et al., 2020).
We evaluated different methods on seven data sets from five experiments that include human mature hepatocytes (Camp et al., 2017), human embryonic stem cells (hESCs) (Chu et al., 2016), mouse embryonic stem cells (mESCs) (Hayashi et al., 2018), mouse dendritic cells (Shalek et al., 2014), and three lineages of mouse hematopoietic stem cells (Nestorowa et al., 2016): erythroid lineage, granulocyte–macrophage lineage, and lymphoid lineage.
These are the same data sets used in Pratapa et al. (2020) and we use their corresponding ground truth networks for our experiments as well. For each data set, there are three versions of ground truth networks: cell-type-specific ChIP-Seq, nonspecific ChIP-Seq, and functional interaction networks collected from STRING. We then have in all 21 different data pairs, 7 different types of expression data evaluated against 3 different types of ground truth.
5.5.1. Preprocessing the real data
For each gene expression data and its corresponding network, we first sorted all the genes according to their variance and select the top 500 varying genes. From the list of known TFs, we only considered all the TFs whose variance had p-value at most 0.01. We then found the intersection between the top 500 varying genes and all the TFs to get a subset of genes that act as the TFs (Table 1). Then, we selected the subgraph of top 500 varying genes from the underlying GRN as our ground truth for evaluation.
5.5.2. Training details
We train on the expression data that is similar to the SERGIO settings for the DS2 data set as it has similar number of genes as the real data. We chose the underlying GRNs for supervision as the random graphs described in Section 5.3. We fixed the number of genes D = 400, the number of cell types C = 9, and total number of single cells M = 2700. The GRNUlar settings were
In general for real data, we observed very low AUPRC values (refer Fig. 6); this is primarily due to the highly skewed ratio between true edges and total possible edges (Davis and Goadrich, 2006; Chen and Mar, 2018). The GRNUlar algorithm clearly outperformed other methods in all test settings. We can further improve the results by tuning the SERGIO simulator settings closer to the real data under consideration. Section 6.1 compares the inference runtimes for various methods.
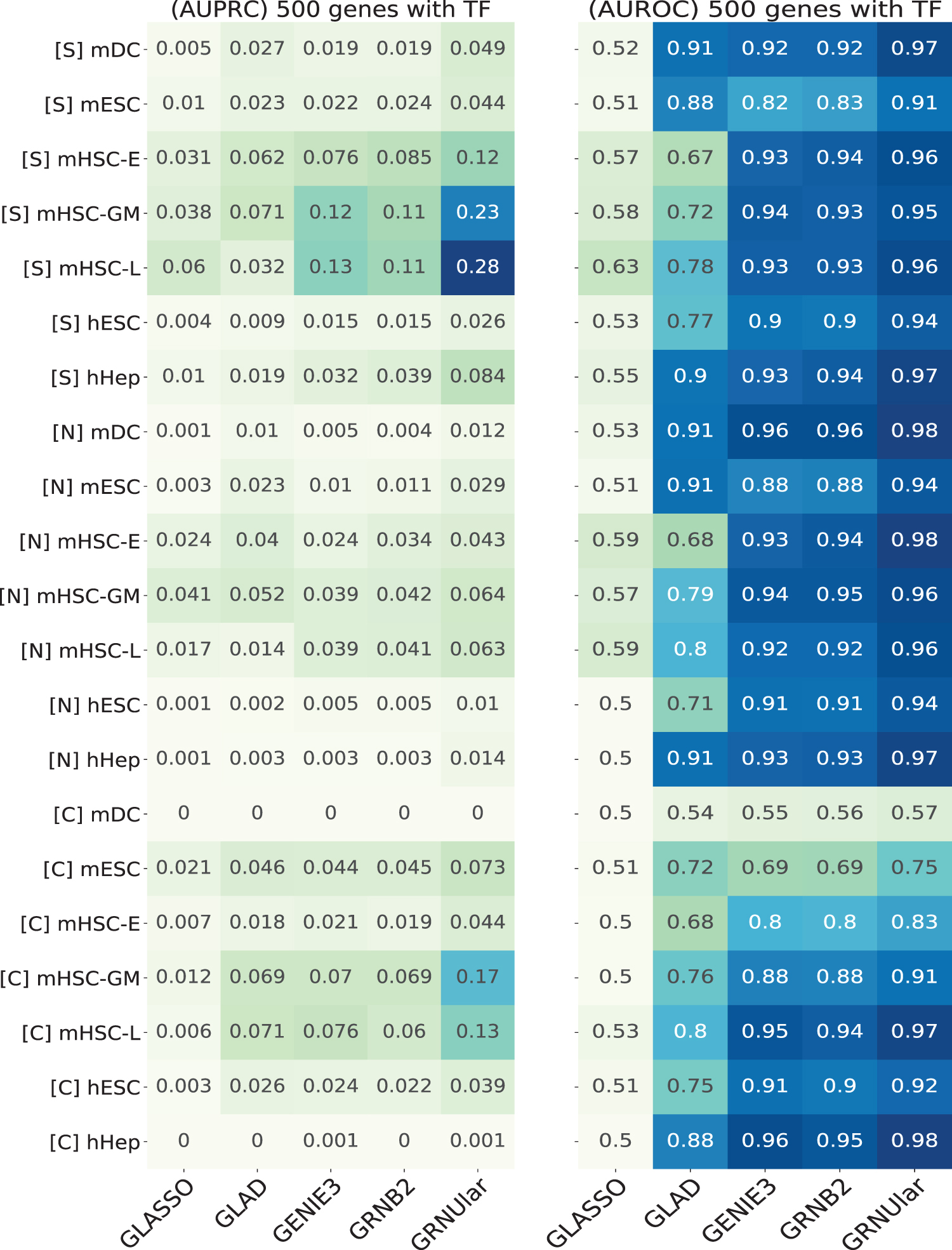
Heatmap of AUPRC and AUROC values of the real data from the BEELINE framework by Pratapa et al. (2020). We ran all the methods including the TF information. [S]/[N]/[C] represent the ground truth networks [String-network]/[nonspecific-ChIP-seq-network]/[cell-type-specific-ChIP-seq] respectively. Data of the species [m] mouse and [h] human were used. GRNUlar performs better than the other algorithms in both the metrics. hESCs, human embryonic stem cells; hHep, human mature hepatocytes; mDCs, mouse dendritic cells; mESCs, mouse embryonic stem cells; mHSCs-E, mouse hematopoietic stem cells-erythroid; mHSCs-GM, mouse hematopoietic stem cells-granulocyte-macrophage; mHSCs-L, mouse hematopoietic stem cells-lymphoid.
6. ANALYZING THE m ESC NETWORK PREDICTED BY GRNUlar
We analyzed the network predicted by GRNUlar from the mESC data set (Hayashi et al., 2018). We chose TFs and genes corresponding to gene ontology terms related to ESC differentiation and cell fate toward endodermal cells as in this data set the ESCs are induced to differentiate into primitive endoderm cells (Hayashi et al., 2018). From BioMart (Kinsella et al., 2011) we obtained 286 genes. We took the intersection between these genes with our predicted GRN (with 500 genes) and found 32 genes.
We first compared the interaction scores predicted by GRNUlar among all 500 genes and the scores among the 32 genes, without applying any threshold. We found that the latter set of scores is significantly higher than the former set of scores (Fig. 7). This means there are more regulatory activities among the genes related to the expected biological processes compared with all the genes selected by variation. We then set the threshold for interaction score as 0.22, and obtained the network shown in Figure 9. In this network, the TFs SOX7, SOX17, MTF2, GATA6, and CITED2 are known TFs in either stem cell differentiation or embryo development; NOTCH1 and RBPJ are TFs in the NOTCH pathway that controls cell fate specification (www.genecards.org). The TFs with highest interaction scores are highly relevant TFs for the cells under study.
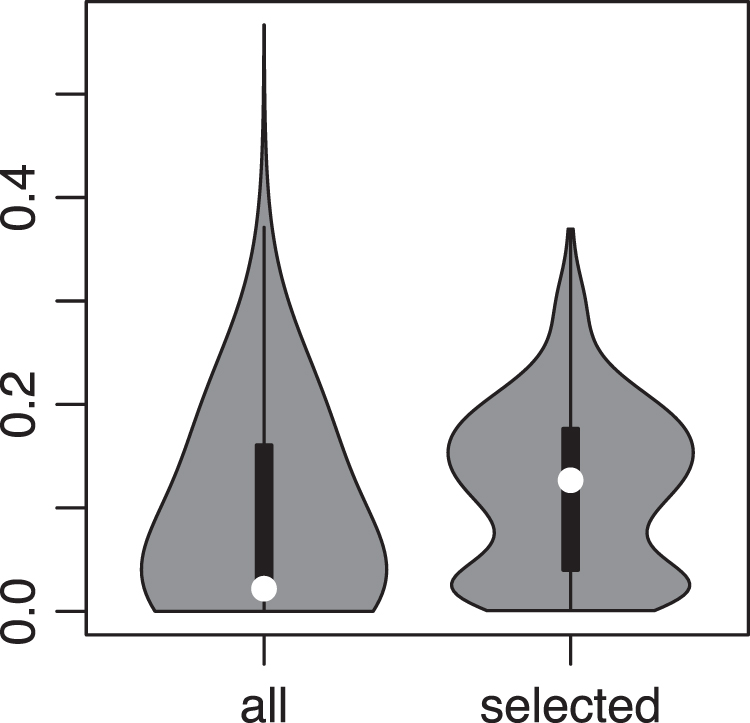
Violin plot comparing the scores of all interactions in the 500 genes (left) and interaction scores between the 32 genes (right). Wilcoxon p-value is 1.3e−14.
We now show how our predicted interactions may bring new biological insights. For instance, we noticed that one of the target genes of SOX7 with strong interaction is CFC1. From ChIP-Seq experiments (the [cell-type-specific-ChIP-Seq] ground truth network mentioned previously), SOX2 is a TF for CFC1.
However, we predicted SOX7 and SOX17 as the TFs for CFC1 in our results. We note that the data set consists of ESCs differentiating into primitive endoderm cells, and SOX2 is a key TF in mESCs governing the pluripotency of the cells (Masui et al., 2007). As the cells differentiate, the pluripotency goes down, so the SOX2 function may also decrease. To verify this, we use the pseudotime of the cells obtained from Pratapa et al. (2020), which was inferred with Slingshot (Street et al., 2018), and visualize the gene expression levels of CFC1, SOX2, SOX7, and SOX17 (Fig. 8). For readability, we plot the actual gene expression levels cell by cell only for CFC1, and for the SOX TFs we plot the fitted lines of their expression levels obtained using LOESS regression. The dashed lines represent the standard deviation.
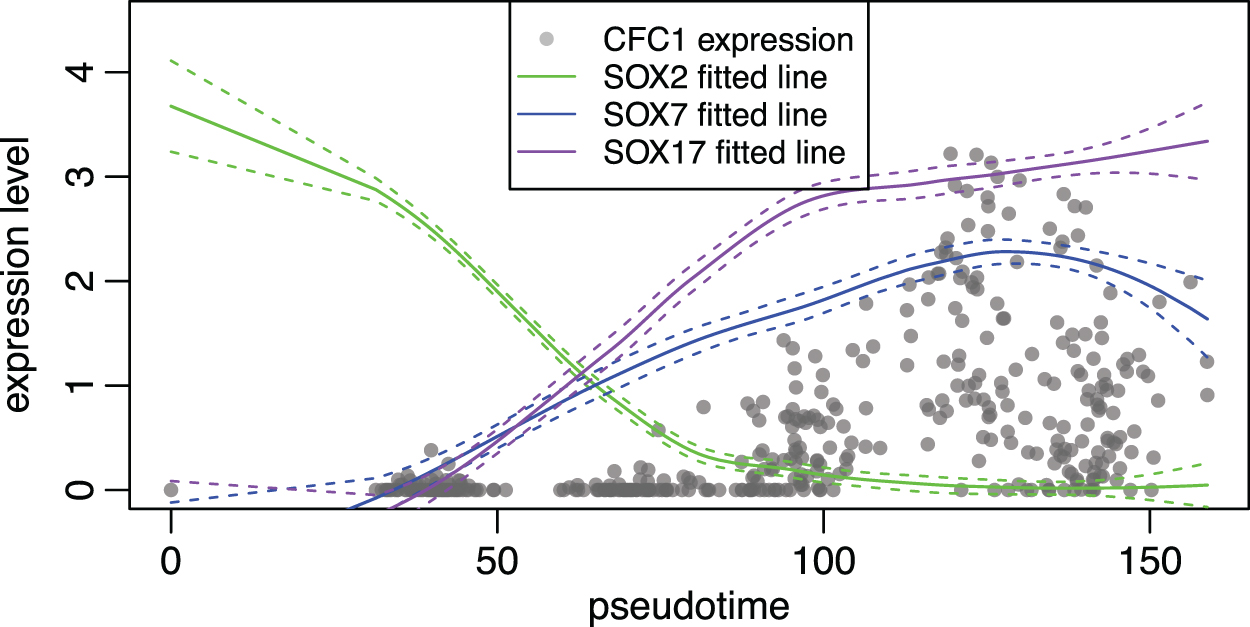
Comparison of gene expression patterns over the pseudotime for CFC1 and the SOX family TFs.
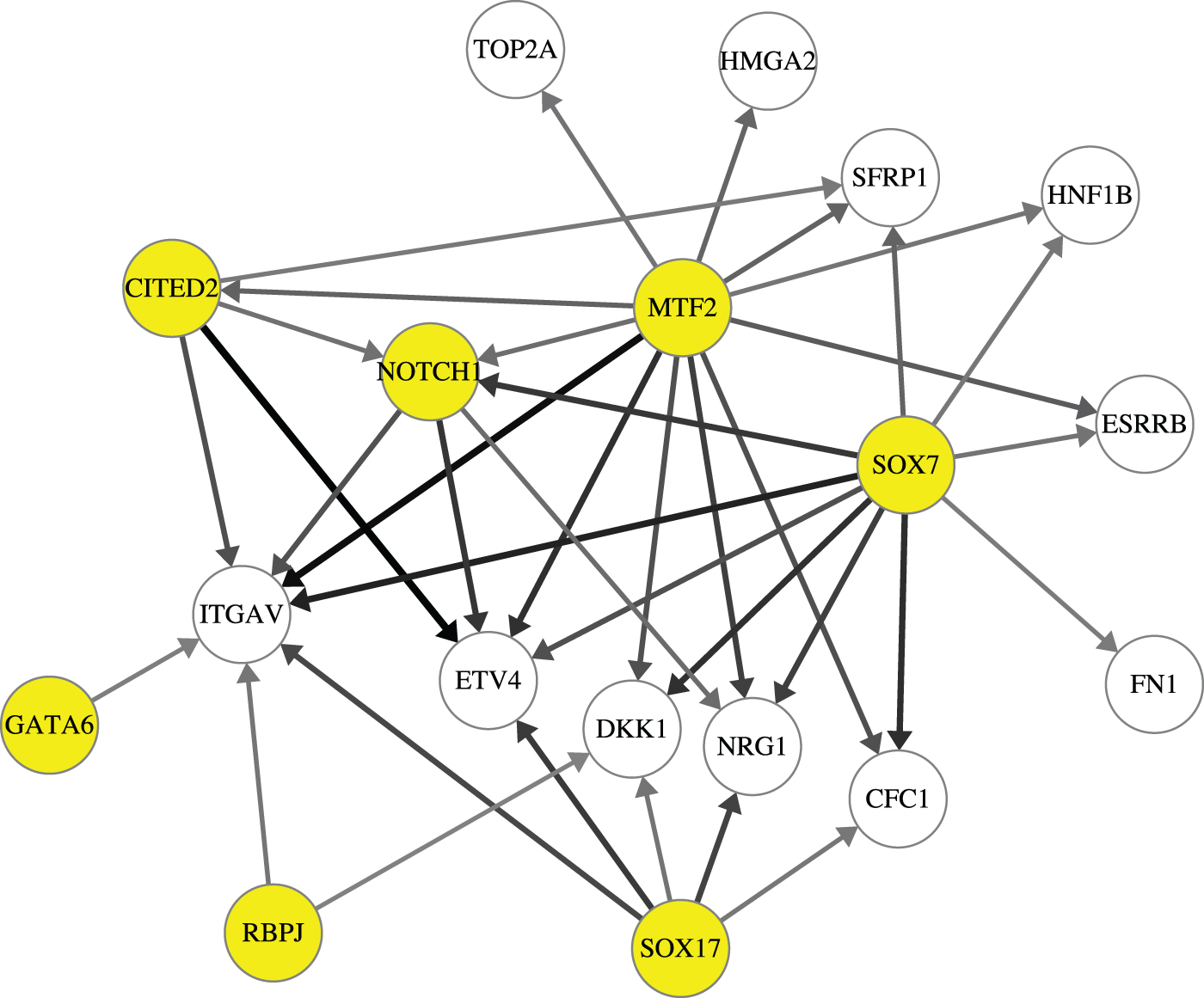
A subnetwork [completed partially directed acrylic graph (CPDAG)] with genes related to stem cell differentiation from GRNUlar predicted network. TFs are the nodes with yellow background. Darker edges mean higher predicted score for the interaction.
We see that indeed the SOX2 expression decreases along the pseudotime, and the expression levels of CFC1, SOX7, and SOX17 increase. The fitted lines of SOX7 and SOX17 show that they are much better predictors for the expression of CFC1 than SOX2. Indeed, it is discussed that SOX7 and SOX17 are highly related members of the SOX family and their high expression in ESCs are correlated with a downregulation of the pluripotency and an upregulation of the primitive endoderm-associated program (Sarkar and Hochedlinger, 2013).
This example showcases how we can use predicted regulatory networks to find regulatory pathways for a specific biological program. Some of these may already have evidence in the literature but some may be new and our prediction can be used to provide hypothesis for further experimental validation.
6.1. Runtimes of different methods
Tables 2 and 3 show the inference time required for different methods with the TF information included. We run different methods on different platforms and hence comparing them directly is not fair. Although we include them to give an idea of the runtimes to the reader.
Inference Runtimes for the GRNUlar Model with Two-Layer Neural Network, As We Vary the Hidden Layer Dimensions Hd
The time is reported for one complete forward call (goodINIT and fitDNN-fast) for D = 1200 genes graph. Other relevant parameters settings were p = 5, L = 15, DNN epochs E1 = 400.
GRNUlar,
Inference Times for Different Methods on D = 1200 Genes Graph
The unrolled algorithms were run on GPUs (NVIDIA P100s), whereas the traditional methods were run on CPU having a single node with 28 cores.
GENIE3; GLAD; GLASSO; GRNB2.
7. CONCLUSIONS AND DISCUSSIONS
We present a deep unrolled supervised learning framework GRNUlar for GRN inference from scRNA-Seq data. The GRNUlar model combines the expressive ability of NNs to capture complex regulation dependencies that manifest in expression data with unrolled learning of sparse graphical models to effectively emulate sparsity of the regulatory networks observed in the real world. We demonstrate that GRNUlar consistently outperforms the representative best-in-class methods on both simulated and real data sets, especially in the more realistic case of added technical noise. Our DL framework accommodates nonlinearity of the regulatory relationships between TFs and genes, and demonstrates high tolerance to technical noise in data.
The unrolled algorithm we proposed is the first supervised DL method for GRN inference from scRNA-Seq data. Our model learns the characteristics of the underlying network through simulated data from GRN-guided simulators such as SERGIO, and demonstrates the novel and effective use of these simulators in training DL models, apart from their traditional use in benchmarking computational methods. Similar techniques can be investigated for other analysis tasks on single-cell data such as clustering and trajectory inference (Luecken and Theis, 2019), by using available realistic simulators for scRNA-Seq data (Dibaeinia and Sinha, 2019; Zhang et al., 2019).
Footnotes
ACKNOWLEDGMENTS
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
This study is supported, in part, by the National Science Foundation under IIS-1841351 and OAC-1828187.