
Abstract
Molecule generation is the procedure to generate initial novel molecule proposals for molecule design. Molecules are first projected into continuous vectors in chemical latent space, and then, these embedding vectors are decoded into molecules under the variational autoencoder (VAE) framework. The continuous latent space of VAE can be utilized to generate novel molecules with desired chemical properties and further optimize the desired chemical properties of molecules. However, there is a posterior collapse problem with the conventional recurrent neural network-based VAEs for the molecule sequence generation, which deteriorates the generation performance. We investigate the posterior collapse problem and find that the underestimated reconstruction loss is the main factor in the posterior collapse problem in molecule sequence generation. To support our conclusion, we present both analytical and experimental evidence. What is more, we propose an efficient and effective solution to fix the problem and prevent posterior collapse. As a result, our method achieves competitive reconstruction accuracy and validity score on the benchmark data sets.
INTRODUCTION
The key challenge of material and drug design is to discover novel molecules that have the desired physical or chemical properties. This process can be understood as an optimization problem as described by Gómez-Bombarelli et al. (2018), and the optimization target is to search for molecules with the optimal desired property scores. However, exhaustive exploration in the molecule space is infeasible since the total number of estimated drug-like molecules is in the order of
As deep learning methods are making more and more achievements in multiple fields as in Miao et al. (2018) and Yang et al. (2020), they have also been applied for molecule sequence generation. The majority of existing molecule generation methods heavily rely on the variational autoencoder (VAE) proposed by Diederik et al. (2014) and Rezende et al. (2014). VAE is the combination of a deep latent variable model and an accompanying variational learning technique. As illustrated in Figure 1, drug molecules can be represented in the simplified molecular-input line-entry system (SMILES) format proposed by Weininger (1988). SMILES is a specification in the form of a line notation for describing the structure of chemical molecules. In Figure 1, the input SMILES sequence
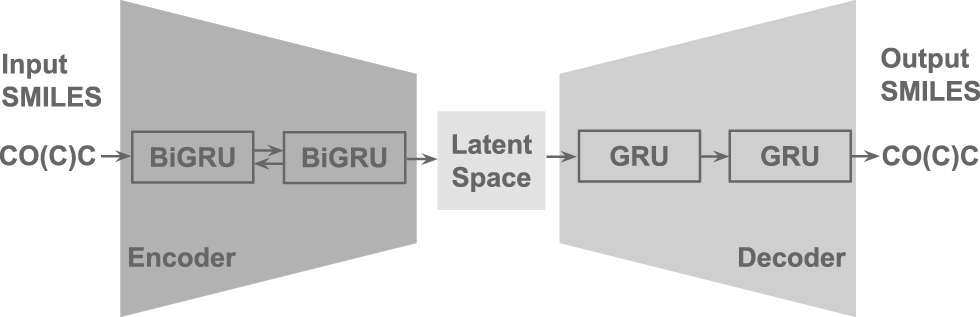
Overview of our VAE model implementation. The encoder and decoder are built based on the bidirectional GRU and unidirectional GRU, respectively. Both the input and output of our model are SMILES sequences. GRU, gated recurrent unit; SMILES, simplified molecular-input line-entry system; VAE, variational autoencoder.
Then, the VAE decoder takes the latent vector as the input to reconstruct the original molecule sequence
However, previous VAE models suffer from the posterior collapse issue, where the decoder tends to ignore latent vectors as described in Bowman et al. (2016) and Gómez-Bombarelli et al. (2018). This problem is more frequently observed in recurrent neural network (RNN)-based models as in He et al. (2019). In consequence, the generated molecules are in low diversity and are hardly relevant to the latent vectors as in Gómez-Bombarelli et al. (2018) and Kusner et al. (2017). This phenomenon has also been observed in natural language processing (NLP) tasks, such as the text generation by Bowman et al. (2016). The major focus of previous NLP-related studies is to propose various training strategies to alleviate this problem, such as the KullbackLeibler (KL) cost annealing by Bowman et al. (2016) and optimizing the decoder multiple times before each encoder update in He et al. (2019).
However, simply extending these methods to molecule generation cannot help molecule generation too much, mostly because the molecule sequences are strictly structured according to SMILES grammar rules and any mutation within the molecule sequences lead to invalid sequences. Motivated by the success of attribute grammars in the compiler design and parse trees in the NLP field, following works such as Kusner et al. (2017) and Dai et al. (2018) propose to incorporate grammar rules to guarantee the validity of generated SMILES sequences. As an alternative, a molecule can also be represented by a graph to avoid the posterior collapse as in Li et al. (2018) and Jin et al. (2018).
Thanks to the development of NLP text generation, the VAE model is applied for molecule generation for the first time in character variational autoencoder (CVAE) by Gómez-Bombarelli et al. (2018). They build a VAE encoder and decoder with GRU layers, representing molecules in the SMILES sequences. However, their model suffers from generating invalid SMILES sequences, which makes their model impracticable. To improve the prior validity, context-free grammars for SMILES are introduced in grammar variational autoencoder (GVAE) by Kusner et al. (2017) to represent a molecule in the sparse tree. However, the validity score is still unsatisfactory. Inspired by this method, syntax-directed variational autoencoder (SD-VAE) by Dai et al. (2018) incorporates extrasemantic rules to ensure that generated SMILES are valid, and it achieves the best performance among all SMILES-based methods. However, these models did not solve the model posterior collapse problem, and there is a large performance gap.
We propose a novel strategy to alleviate the posterior collapse problem considering the essential drawbacks of the contemporary RNN-based VAE models in the molecule generation situation. To achieve this goal, we carefully analyze the posterior collapse problem of the vanilla VAE model for SMILES sequence generation. We point out that the underestimated reconstruction loss triggers the posterior collapse issue in the molecule sequence generation, as the direct consequence of the imbalance between reconstruction loss and KL loss during VAE training.
To overcome the problem, we propose a novel loss function to leverage the trade-off between the reconstruction loss and the KL loss in VAE training. Without modifying the VAE network structures or costing extracomputational complexity, our proposed strategy is extremely simple yet effective in preventing the posterior collapse in molecule generation. We also provide a detailed analysis of our method* and empirically demonstrate its excellent reconstruction accuracy and competitive validity score on the ZINC 250K data set from Kusner et al. (2017) and on the GuacaMol data set from Brown et al. (2019).
This article is a major extension of our previous conference version (Yan et al., 2020b). In addition to the experimental verification for the statement that the underestimated reconstruction loss causes the posterior collapse of the VAE models, we also provide theoretical analysis and proof in this work. Besides, to further improve the validity score of our method, we introduce a partial SMILES sequence check toolkit PartialSmiles † to verify the validity of the SMILES sequence during the molecule generation process. What is more, to better evaluate the proposed method, we include the results of two extra evaluation metrics novelty and uniqueness in experimental comparison with baseline methods. Last but not least, we conduct experiments on the extra large-scale data set GuacaMol, which consists of 1.6 M molecules to demonstrate the scalability and generalization of our proposed method.
The variational autoencoder
The VAE is a specially regularized variant of the standard autoencoder (AE). It is appealing because it can learn complex distribution in an unsupervised manner and later can act as a generative model defined by a prior distribution
where the VAE encoder
The VAE training is optimized to maximize the ELBO, where (1) negative reconstruction loss
The posterior collapse phenomenon has also been reported in previous works on NLP text generation such as Bowman et al. (2016), Yang et al. (2017), and Kim et al. (2018). When posterior collapse happens, the model training falls into the local optimum of the ELBO, in which the decoder tends to ignore z when training the VAE model and the variational posterior
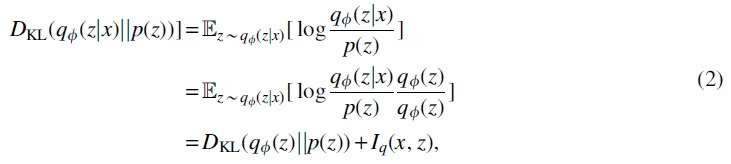
where
When posterior collapse occurs, the KL loss decreases nearly to zero so that Iq is also close to zero [both items on the right-hand side in Equation (2) are non-negative] during the VAE model training process. It is especially evident when modeling discrete data with a strong autoregressive network such as long short-term memory (LSTM) by Hochreiter and Schmidhuber (1997) and GRU by Chung et al. (2014), which is exactly our case for molecule sequence generation. This is undesirable since the VAE model fails to learn meaningful latent representations for input molecule sequences.
For text generation task in NLP, the posterior collapse problem has been mainly attributed to the low quality of latent representations z at the early stage of model training as pointed out by Bowman et al. (2016), He et al. (2019), and Fu et al. (2019). To be more specific, the decoder
However, molecule SMILES generation is a quite different scenario, although it appears to be same as the NLP text generation. First of all, its vocabulary size is far less than the NLP text generation data sets. The token size of NLP text data is usually tens of thousands or even larger, while it is less than 100 for chemical molecule data. The smaller token size makes the molecule reconstruction task much easier. Second, the molecule sequence is composed strictly following the SMILES grammar rules, and the reconstructed sequence must be exactly the same as the input to be matched successfully. Any token mutations can result in an invalid sequence. However, there are no such rigid grammar rules applied to the NLP text, and the exact match is not required.
We find that the existing solutions of He et al. (2019) and Fu et al. (2019) for NLP text generation performs poorly in the chemical molecule generation. This motivates us to propose such a solution for molecule sequence generation.
To investigate the cause of the posterior collapse in the VAE for molecule sequence generation, we conduct extensive analysis and investigation into the posterior collapse process. We find it is the underestimated reconstruction loss that causes posterior collapse during VAE training process. Both theoretical analysis and experimental support are provided to verify our hypothesis.
The reconstruction loss term
We can rewrite the reconstruction loss term in Equation (1) as:
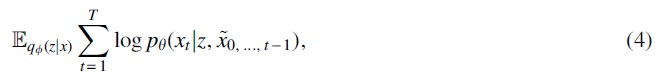
where the T is the maximum time step,
With teacher forcing training method, now the actual reconstruction loss during VAE training is:
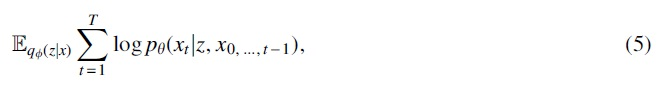
where
We posit

The ground-truth information
Rebalanced VAE loss
Since reconstruction loss is underestimated during training and it breaks the balance with KL loss, which eventually leads to the posterior collapse, we propose to recover the balance by applying a weight
where
Inspired by the
Note that in our case
Except for the above analysis, our method can also be explained from an intuitive perspective. In previous methods CVAE, GVAE, and SD-VAE, when sampling latent vectors z they have to reduce the standard deviation
We acknowledge that previous methods such as Dai et al. (2018), He et al. (2019), and Fu et al. (2019) have empirically tried to reduce the KL loss weight to avoid the posterior collapse. The
Our proposed solution to the VAE model posterior collapse is simple but extremely effective and efficient. We do not need to modify the network architecture and only adjust the training loss slightly, without introducing much extra computation. In this section, we will first train a vanilla VAE model and track the process of model collapse, as well as experimentally verify that the reconstruction loss is underestimated. Then, we will conduct extensive experiments to demonstrate the effectiveness of our proposed method.
Experimental settings
We build our VAE model based on GRU layers. The VAE encoder is composed of two layers of bidirectional GRU, which is good at capturing sequence representation as Schuster and Paliwal (1997), and hidden size of each GRU layer is 512. The decoder is made up of four layers of unidirectional GRU with the same hidden size of 512. Following previous works (Gómez-Bombarelli et al., 2018; Jin et al., 2018), we use unit Gaussian prior and set the latent vector dimension to be 56. The ELBO objective is optimized with Adam optimizer by Kingma and Ba (2014) and learning rate is 0.0001. The model is trained with teacher forcing and KL loss annealing. We train the model for 150 epochs and report the performance of the final model. Experiments are conducted on a machine with an Intel Core i7-5930K@3.50GHz CPU and a GTX 1080 Ti GPU.
We experiment on the ZINC 250K data set by Kusner et al. (2017), which is a subset of the ZINC by Sterling and Irwin (2015). Molecule sequences are tokenized with the regular expression from Schwaller et al. (2018). We use the same training and testing split as previous works (Kusner et al., 2017; Jin et al. 2018) and have 10K hold-out data out of the training as the validation data. We also experiment on a large-scale data set GuacaMol by Brown et al. (2019), which is derived from the ChEMBL 24 database by Mendez et al. (2019) to demonstrate the scalability and generalization of our method. GuacaMol data set consists of 1.6M molecules, and we adopt the same data split provided by Brown et al. (2019). We will use the same experimental settings in all our experiments unless explicitly stated.
As for the model evaluation metrics, we report the reconstruction accuracy, validity, novelty, and uniqueness scores following previous work. Following Jin et al. (2018), we encode each molecule from test data set and then decode obtained latent vector to reconstruct input molecule SMILES. The reconstructed SMILES must be exactly the same as the input to be counted as successful. The reconstruction accuracy is defined to be the ratio of successfully reconstructed molecule sequences to the total tried reconstruction. To calculate validity, 10K latent vectors are randomly sampled from the prior distribution as the input for the decoder.
The validity is the portion of chemically valid reconstruction SMILES from the random sampling to the total decoded sequences. We use open-source tool RDKit by Landrum et al. (2006) to check the validity of SMILES. The novelty is the ratio of generated chemically valid molecules, which are not present in the training data set to the total generated chemically valid molecules. It evaluates the model's ability to generate novel molecules. The uniqueness is used to evaluate to what extent a model generates unique chemically valid molecules, and it is defined as the ratio of generated chemically valid molecules that are unique.
VAE training dynamics
We track the training process of a vanilla VAE model, as well as that of our proposed method. We investigate training dynamics including the KL loss weight, KL loss, reconstruction loss, mutual information, reconstruction accuracy, and validity score. Mutual information
which is actually the same as Equation (2). We approximate the aggregated posterior
As a comparison, we also illustrate the training dynamics of our proposed method. We set KL weight
Results of the two models' training are plotted in Figure 2. The vanilla VAE model performs well on the validation data at the early stage of the KL weight annealing. However, as the KL weight increases, KL loss drops quickly as expected since more penalty is added to the KL loss term, whereas the reconstruction loss starts to rise at the same time. The mutual information

Training dynamics of vanilla VAE model and our method on validation data. We track
However, our method achieves lower reconstruction loss early and can maintain it during model training. Although the KL loss of our method is larger than the vanilla VAE, considering that we have a smaller KL weight
We find that introducing ground-truth information into the decoder will result in underestimated reconstruction loss in the previous section and have provided our detailed analysis previously. In this section, we will experimentally verify that the reconstruction loss is indeed underestimated during the training. We can estimate how much the reconstruction loss has been underestimated using Monte Carlo sampling. Specifically, we can sample a batch of data and then run the model with and without the teacher forcing, respectively. The underestimated ratio can be approximated by the ratio of reconstruction loss with teacher forcing to that without teacher forcing.
We track the reconstruction loss on the validation data set when the teacher forcing is applied and removed, respectively. Results are shown in Figure 3a. When teacher forcing is applied, the reconstruction loss drops close to 1 quickly, whereas the loss is much larger (at least 7.5) without teacher forcing. This is not unexpected since the prediction error may be accumulated during the decoding process without teacher forcing. Any wrong token prediction as RNN input at the next time step may result in the following prediction totally different from ground-truth sequences.

To quantitatively evaluate how much the reconstruction loss has been underestimated, we can compute the ratio as reconstruction loss with teacher forcing to that without teacher forcing at each time step. Results are shown in Figure 3b. It confirms our conclusion that the reconstruction loss is underestimated. To recover a rebalanced VAE loss, we can set KL loss weight exactly as the underestimated ratio in each epoch. But this requires us to compute the ratio repetitively during training, which is time-consuming. To be simplified, we set
We summarize the molecule reconstruction accuracy, validity, novelty, and uniqueness scores on the ZINC 250K test data in Table 1. Our method outperforms all previous models in reconstruction accuracy by a large margin (16% larger than the second-best model). In the meanwhile, our method achieves 90.7% validity, which is much better than previous SMILES-based methods. We can further boost our model performance by incorporating a SMILES validating parser PartialSmiles, which can check the validity of the SMILES prefix easily when generating SMILES sequences token by token. The validity score can be boosted to 93.8% with PartialSmiles.
Reconstruction Accuracy and Validity Results on the ZINC 250K Data Set
Reconstruction Accuracy and Validity Results on the ZINC 250K Data Set
Bold value in column 3 is the best result.
Baseline results are reported in Kusner et al. (2017), Dai et al. (2018), Simonovsky and Komodakis (2018), and Jin et al. (2018).
The SMILES validating parser PartialSmiles is applied during the generation. The novelty and uniqueness scores of baseline methods are copied from Samanta et al. (2020).
CVAE, character variational autoencoder; GVAE, grammar variational autoencoder; JT-VAE, junction tree variational autoencoder; VAE, variational autoencoder; SD-VAE, syntax-directed variational autoencoder; SMILES, simplified molecular-input line-entry system.
Compared with other SMILES-based methods, our model is much superior in both the reconstruction accuracy and prior validity, even if complex grammar or syntax rules are incorporated in Kusner et al. (2017) and Dai et al. (2018). Note that the junction tree variational autoencoder (JT-VAE) model assembles molecules by adding subgraphs step-by-step to make sure that the generated molecule graphs are always valid. However, these subgraphs are extracted from the training data set, which limits the JT-VAE from generating molecules with unseen subgraphs.
While our method achieves competitive validity performance without any constraints and is able to generate novel molecules that are not from the same distribution as the training data. That is one important reason why our method achieves better reconstruction accuracy, whereas JT-VAE suffers from reconstructing testing molecules (Mohammadi et al., 2019). Besides, our method is much more efficient than JT-VAE. When generating 10,000 unique valid SMILES from prior random sampling, JT-VAE ‡ (faster version) takes about 1450 seconds while our method only needs 9 seconds.
As for the novelty and uniqueness, our method achieves 100.0% for both metrics, which are the same as other SMILES-based methods including GVAE and SD-VAE. Note that the novelty and uniqueness are evaluated only on the chemically valid molecules. This indicates that even if both GVAE and SD-VAE achieve the same novelty and uniqueness scores, our method can generate much more valid molecules than GVAE and SD-VAE due to the better validity score. JT-VAE achieves only 99.9% novelty score and 99.1% uniqueness score. This demonstrates that our model is a better chemically valid molecule generator.
We also experiment on a large-scale data set GuacaMol to evaluate the scalability and generalization of our proposed method and report results in Table 2. We use the same experimental settings as the ZINC 250K data set. Our method achieves 92.6% reconstruction accuracy, 90.6% validity score, 100.0% novelty, and 100.0% uniqueness on the GuacaMol data set, which are similar to the performance on ZINC 250K. By checking the validity of SMILES during the generation, the validity score can be further boosted to 93.6%. The experiment on the large-scale data set demonstrates our method scales and generalizes well on a large data set.
Reconstruction Accuracy and Validity Results on the GuacaMol Data Set
The SMILES validating parser PartialSmiles is applied during the generation.
Our model achieves 92.7% reconstruction accuracy, and all reconstructed SMILES are valid on the ZINC 250K data set. We investigate the reconstruction results further and find that our model can predict 97.3% of all tokens correctly, which is measured at the level of the token instead of the sequence. Besides, most of the unmatched sequences (62%) are valid, and it confirms the reconstruction ability of our model. We show some valid but unmatched examples in Figure 4. Even for these unmatched examples, there is only a small ratio of the predicted tokens that are different from the ground-truth, which demonstrates the reconstruction ability of our method.

Reconstruction error examples. Unmatched tokens between the input and reconstruction SMILES are highlighted. Note that “[O-]” is a single token.
As for the validity sore, we also investigate the model outputs. We find that our model can generate complicated and diverse molecules with multiple rings. As for the invalid sequences, from both the reconstruction and prior sampling, there are several typical errors: (1) unkekulized atoms, (2) valence error, (3) unclosed ring, and (4) parentheses error. We believe that the grammar-based methods of Kusner et al. (2017) and Dai et al. (2018) are complementary to our method and can be combined together to reduce these errors.
One of the important tasks in the drug molecule generation is to make molecules with desired chemical properties. We follow Kusner et al. (2017) and Jin et al. (2018) for all the experimental setting, and the optimization target score is:
where
We first associate each molecule with a latent vector, which is the mean of the learned variational encoding distribution. The latent vector for each molecule will be treated as its feature, and we train a sparse Gaussian process (SGP) to predict the target score
We report the SGP prediction performance when trained on latent representations learned by different models. We train the SGP with 10-fold cross-validation and report the top-3 molecules found by the BO. As shown in Table 3, molecules found by our model are much better than that found by previous SMILES-based methods, and our method is even superior to the graph-based method JT-VAE. Figure 5 shows the top-3 molecules found by our model.
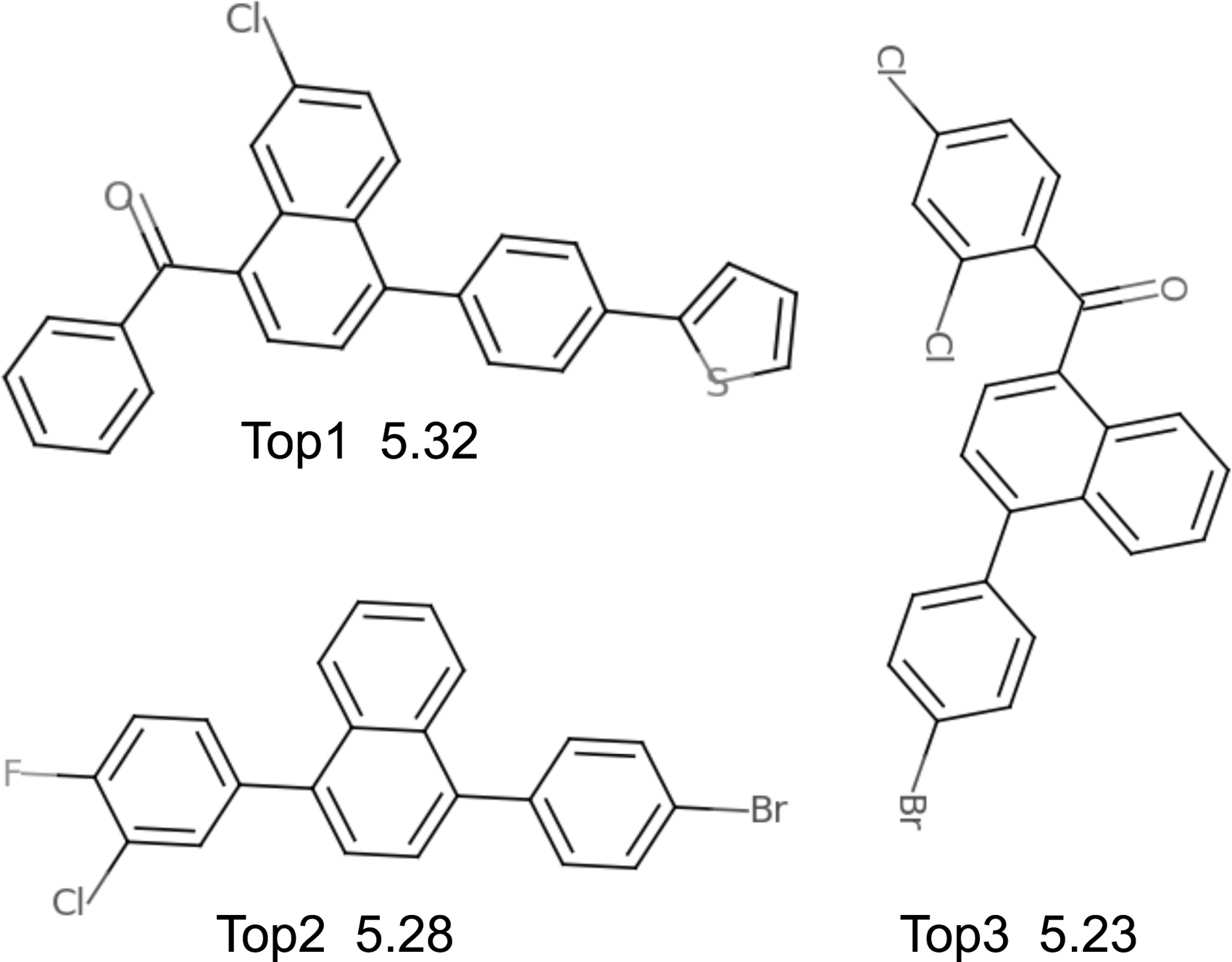
Top-3 molecules and associated scores found by our model with Bayesian optimization.
Top-3 Molecule Scores Found by Bayesian Optimization
Our method is very efficient, and it works extremely well in the molecule generation, in which SMILES sequences are highly structured and grammarly organized. Our experimental results indicate that grammar and syntax rules are necessary to generate more valid SMILES sequences, and they are complementary to our method. Besides, SMILES-based methods and graph-based methods may also be combined together to boost the model performance further.
Although our primary focus is the VAE for molecule generation, our method can also help the NLP task as we mentioned at the end of Section 2.4. Reducing KL loss weight can help the VAE model for the NLP task avoid the posterior collapse as shown in He et al. (2019) and Fu et al. (2019).
The latent representation learnt by our model can be applied to various downstream tasks, such as molecule property prediction (Xu et al., 2017; Ma et al., 2020, 2021, 2022). In the future, we may explore more about this application.
CONCLUSIONS
In this work, we investigate the posterior collapse problem in VAE for molecule sequence generation. Through extensive analysis, we conclude that the underestimated reconstruction loss results in the posterior collapse. The conclusion is supported by both theoretical analysis and experimental results. Based on our analysis, we propose a simple and effective solution to overcome the underestimated reconstruction loss problem by weighting the KL loss term. With the proposed rebalanced VAE loss, the VAE model can avoid the posterior collapse problem and achieve excellent performance in both reconstruction accuracy and validity score on two data sets. We also demonstrate the excellent generalization of our method on a large-scale data set.
Footnotes
AUTHORS' CONTRIBUTIONS
C.Y.: Conceptualization, methodology. J.Y.: Formal analysis. H.M.: Writing—review and editing. S.W.: Resources. J.H.: Supervision.
ACKNOWLEDGMENT
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
This work was partially supported by the U.S. National Science Foundation IIS-1553687 and Cancer Prevention and Research Institute of Texas (CPRIT) award (RP190107).