
Abstract
Genome-wide association studies (GWASs) are often confounded by population stratification and structure. Linear mixed models (LMMs) are a powerful class of methods for uncovering genetic effects, while controlling for such confounding. LMMs include random effects for a genetic similarity matrix, and they assume that a true genetic similarity matrix is known. However, uncertainty about the phylogenetic structure of a study population may degrade the quality of LMM results. This may happen in bacterial studies in which the number of samples or loci is small, or in studies with low-quality genotyping. In this study, we develop methods for linear mixed models in which the genetic similarity matrix is unknown and is derived from Markov chain Monte Carlo estimates of the phylogeny. We apply our model to a GWAS of multidrug resistance in tuberculosis, and illustrate our methods on simulated data.
INTRODUCTION
Genome-wide association studies (GWASs) are designed to identify the genetic variants affecting phenotypes of interest such as multidrug resistance in tuberculosis (MDR-TB) (Price et al, 2006; Zhang et al, 2010). Classic approaches to GWASs rely on linear association tests to quantify the relationship between phenotypes and genotypes.
Population structure (Patterson et al, 2006) in the phylogeny of bacterial genomes can lead to false positives, spurious associations, or inflated p values (Novembre et al, 2008). The genealogy of tuberculosis (TB) typically exhibits strong clade structure (Cordero and Polz, 2014; Earle et al, 2016), with geographically widespread lineages, and so GWASs on TB are vulnerable to population stratification.
Linear mixed models (LMMs) use the genetic similarity among samples as a random effect. This controls for confounding from population structure, leading to improved false discovery rates (FDRs). In Kang et al (2010), the efficient mixed-model association (EMMA) expedited was proposed, which computes the variance component in linear mixed models in an efficient way. In addition, factored spectrally transformed linear mixed models (FaST-LMMs) were introduced (Lippert et al, 2011; Listgarten et al, 2013), with running time and memory costs that scale linearly in the cohort size. In Zhou and Stephens (2014), Yang et al (2011), and Dahl et al (2016), models were developed for computationally efficient linear mixed effects with multivariate phenotypes.
The efficiency of FaST-LMM methods has been further improved by subsetting the genetic variants examined, so that a set of maximally independent genetic variants is considered (Listgarten et al, 2012). Several other methods have been proposed to scale to large cohorts (such as U.K. Biobank; Bycroft et al 2018; Sudlow et al 2015). Loh et al (2015) developed an efficient Bayesian mixed model, implemented in the software BOLT-LMM, that requires lower computational costs than standard LMMs, while increasing power by modeling genetic architectures through a Bayesian mixture prior on the effect sizes of the genetic variants.
Loh et al (2018) also proposed a much faster version of BOLT-LMM and demonstrate the method by analyzing the U.K. Biobank data. Jiang et al (2021) developed generalized linear mixed model (GLMM)-based methods for GWASs (fastGWA-GLMM) for binary phenotypes. The method is scalable to cohorts with millions of individuals.
All the mentioned LMM methods assume that the matrix specifying the genetic similarity among the samples is known (i.e., through an empirical genetic similarity matrix, Patterson et al 2006; or a kinship matrix derived from a pedigree, Kirkpatrick et al 2019). For large cohorts of human genotypes, there is often low uncertainty about estimated genetic similarity matrices.
However, for some studies, such as bacterial studies, in which small number of samples or loci is present, or for studies in which genotyping is sparse and noisy, uncertainty about the genetic similarity matrix may degrade the quality of LMM results (e.g., in Wang et al, 2021, heritability estimates based on genetic similarity matrices were found to have large variance, which may translate into reduced power for LMMs conducted with point estimates of the genetic similarity matrix).
In the pyseer (Lees et al, 2018) package, a few methods for GWASs are implemented. For example, a fixed effects model using the genetic similarity matrix represented by a multiple-dimensional scaling (MDS) approximation, a linear mixed model using a kinship matrix, and a whole genome model using elastic net. However, the genetic similarity matrix represented by MDS is not equal to its expectation (Patterson et al, 2006) and is biased (Wang et al, 2021).
MDR-TB is a major concern for TB control (Grandjean et al, 2017). MDR in TB is caused by genetic variations in genes that encode drug targets and drug-converting enzymes (Coll et al, 2014). Understanding these effects is critical for improving treatment for MDR-TB patients. But population stratification (in which genetic variates correlate with structure in geographical or socioeconomic indicators) and noisy genotyping of bacterial genomes confound such studies (Price et al, 2006; Zhang et al, 2010). In this study, we improve the control provided by linear mixed models by encoding uncertainty about genetic similarity, and report applications on TB data and simulated data.
We propose a new LMM method for GWASs, using phylogenetic trees to control for population structure. We use Markov chain Monte Carlo (MCMC) to draw samples for the phylogeny based on observed genetic sequences, and then we compute the expected genetic similarity matrix induced by each phylogeny (Wang et al, 2021). We then apply the linear mixed model to each sampled expected genetic similarity matrix and average the results.
Simulations show that the true positive rates (TPRs) and FDRs of our method outperform both existing linear regression methods, LMM methods in which the genetic similarity matrix is estimated empirically, and pyseer with the genetic similarity matrix represented using consensus tree of MCMC posterior samples. We apply this method to MDR-TB in a GWAS of 467 TB subjects in a population from Lima, Peru (Grandjean et al, 2017).
2. METHODS
Linear mixed effects models for GWASs
We consider a population of samples typed at given single nucleotide polymorphisms (SNPs) and with measured phenotypes. We begin this section with an exposition of the linear mixed model (Kang et al, 2010; Lippert et al, 2011). Let the study subject indices be
The LMM is a mixed effects model for association between SNP Gm and the phenotype. Independent LMMs may be applied at each SNP as follows:
Here

Here
Although previous LMM study approximates
The covariance matrix of the random effects for the m-th SNP is the positive-definite matrix
In this study, we propose a new linear mixed model for multivariate GWASs, by assuming an unknown genetic similarity matrix
Here we consider estimating the phylogeny t in a Bayesian framework. We place a proper prior distribution on the phylogenetic tree t (i.e., a uniform clock prior for a binary clock tree). After specifying the prior distributions, trees can be sampled conditioned on genotype data using standard software packages for phylogenetic inference (e.g., MrBayes; Ronquist et al 2012).
When multiple posterior samples of the phylogeny
We associate each posterior sample
Here
We note that

The computational cost for reconstructing a tree through MCMC is a linear function of
For the experiments on TB data from peru, the sample collection and processing received ethical approval from the IRB of the Universidad Peruana Cayetano Heredia. In addition, the TB sequences are available through the European Nucleotide Archive (accession no: PRJEB5280) and are in the public domain.
Simulated data sets
Simulation 1
In the first simulation study, we simulated data sets for four scenarios: A, B, C, and D. The binary trees were simulated through the ms software (Hudson, 2002). We used the R package phangorn (Schliep, 2011) to create genetic variants under the assumption of the Jukes Cantor model (Jukes and Cantor, 1969). We assumed that the branch lengths are in units of
We hypothesize that with a larger number of markers, the genetic similarity matrix will have less uncertainty, and LiMU results may approach those of the LMM. In scenario A: small data set, we simulated 50 trees with
In scenarios C: Illumina, and D: Nanopore, we simulated the phenotype through the linear model (LM) with
We compared the TPR and FDR of LiMU, LMM (using the empirical genetic similarity matrix for the kinship), the fixed effects model with the genetic similarity matrix represented by MDS implemented in the pyseer software, a linear mixed effects model with the expected genetic similarity matrix computed from consensus tree of MrBayes (CLMM), and a linear model in a task in which associated genetic variants are recovered. To compute the similarity matrix of the model implemented in pyseer for controlling population structure, we used the consensus tree provided by MrBayes.
The estimation of linear mixed model was carried out using the efficient mixed model association (EMMA; Lippert et al 2011). For a fixed p value threshold
Figure 1 displays the ROC found for these methods for data sets A: small data set (upper left panel), B: GCTA model (upper right panel), C: Illumina (middle left panel), and D: Nanopore (middle right panel). The lower panels show the partial ROC curves of the top left corner for scenarios B, C, and D. In scenarios A and B, the ROC curve of LiMU dominates those of pyseer, LMM, and LM at all FDR levels in both scenarios. In scenario C, the data were generated through a linear regression model, the ROC curves found for all methods were close. In scenario D, with higher genotyping error in data simulation, the area under all ROC curves was lower (compared with scenario C).

ROC provided by LM, LMM, the FIXED, a linear mixed effects model with the expected genetic similarity matrix computed using the consensus tree of MrBayes according to Wang et al (2021), and a LiMU for data sets
The area under the receiver operating characteristic (ROC) curve (AUC) and the improvement at FDR = 0.05 are listed in Table 1. Figure 2 shows the TPR at a fixed FDR level 0.05 for the four scenarios shown in Figure 1. The ROC curves provided by LiMU and LMM with the expected genetic similarity matrix computed from the consensus tree according to Wang et al (2021) exhibit similar performance.
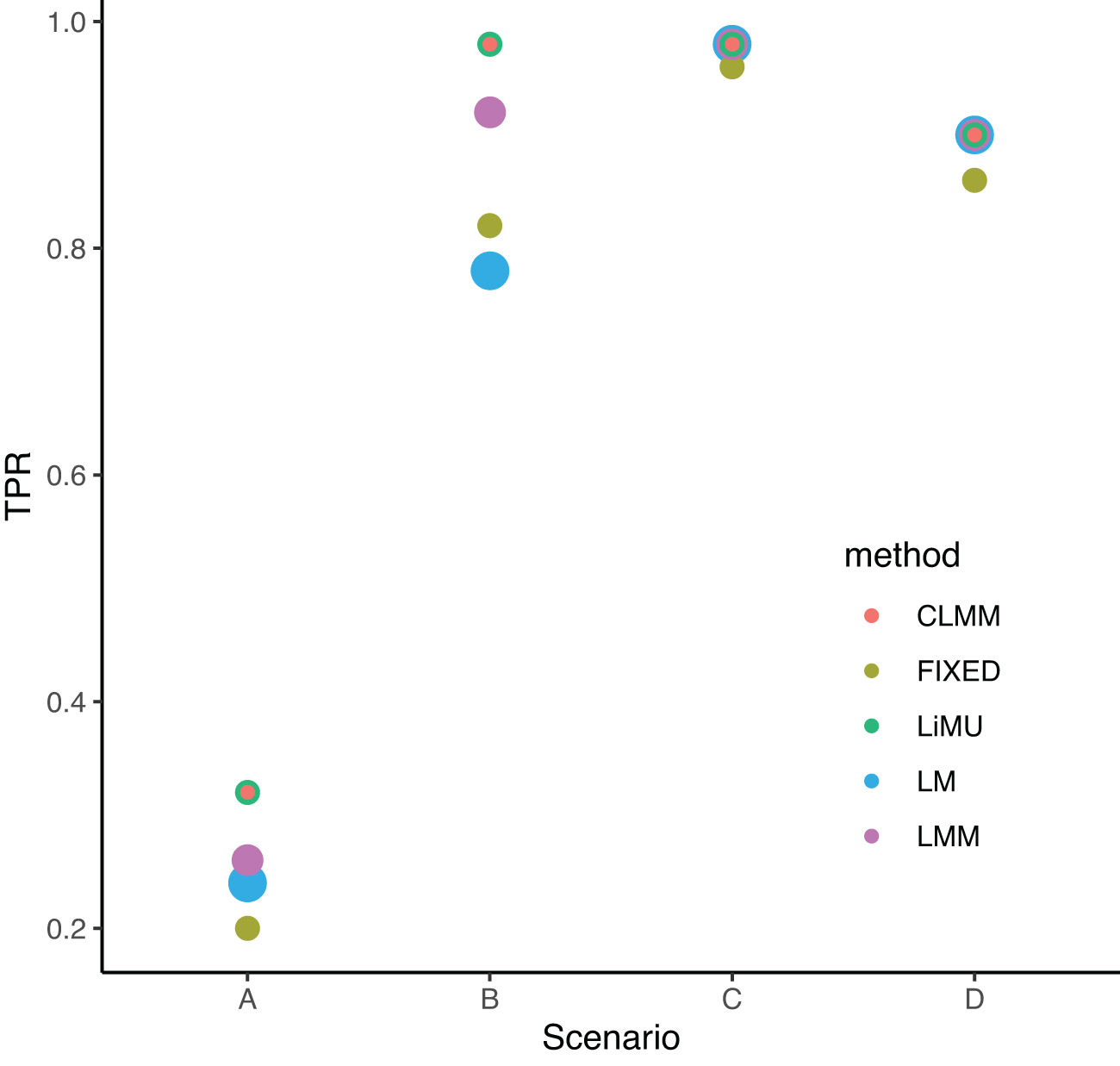
TPR at a fixed FDR level
AUC and True Positive Rate at False Discovery Rate 0.05 for Simulation Scenarios A, B, C, and D
LiMU shows improved AUC and TPR in scenarios A and B. The AUC and TPR are close for all methods in scenario C.
AUC, area under the ROC curve; CLMM, consensus linear mixed model; FDR, false discovery rate; FIXED, fixed effects model implemented in the pyseer software; GCTA, Genome-wide Complex Trait Analysis; LiMU, linear mixed model with uncertain genetic similarity matrix; LM, linear model; LMM, linear mixed model; TPR, true positive rate.
Bold indicates the best method (highest TPR or highest AUC).
In the second simulation study, we first examined the area under the ROC curve (AUC) provided by the four methods already discussed as a function of
We also report the compute time required for each step of LiMU in the first row of Table 3. The experiments were conducted on a 2.3 GHz Intel Core i9 processor. Half a million iterations of MrBayes run costs
We examined the area under the ROC curves (AUC) induced by the p values of linear regression (LM), linear mixed model with empirical genetic similarity matrix (LMM), the pyseer software (pyseer) and a linear mixed model with unknown genetic similarity matrices (LiMU), for simulated data in which the ground truth was known. Figure 3 displays the area under curve (AUC) as a function of
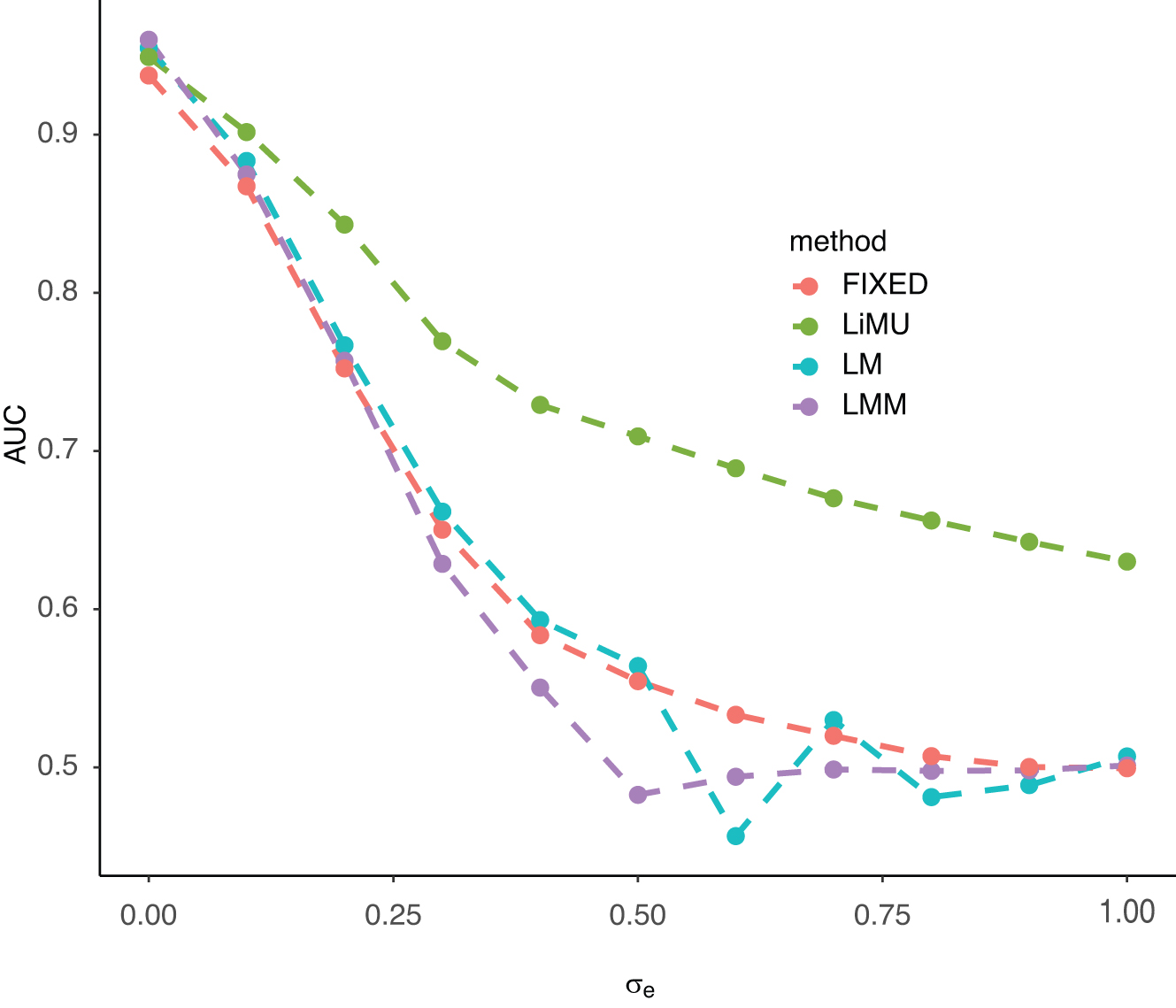
AUC as a function of
In addition, we examined the area under curve (AUC) as a function of

AUC as a function of
In the third simulation study, we design experiments with more sophisticated setups. We create 50 data sets, in each of them we randomly sample
Figure 5 shows the density plot for the first two components provided by metric MDS (Williams, 2000). The MDS is conducted on the pairwise distance between the 100 thinned posterior samples for one of the 50 data sets using treespace.

The density plot for the first two components provided by metric MDS. The MDS is conducted on the pairwise distance between the 100 thinned posterior samples for 1 of the 50 data sets using treespace. MDS, multidimensional scaling.
We investigate the effects of polygenic traits using LiMU, linear model (LM), linear mixed model with empirical genetic similarity matrix (LMM), and the fixed effects model implemented in the pyseer software (FIXED). The genetic sequences are obtained by randomly sampling M sites from the original TB sequences. We examine three levels of M,

where
Figure 6 shows the AUC as a function of M provided by LiMU, linear model (LM), linear mixed model with empirical genetic similarity matrix (LMM), and the FIXED in four scenarios with size of effects 1 (upper left), 2 (upper right), 5 (lower left), and 10 (lower right). The AUCs provided by LMM and LiMU are higher than those provided by LM and FIXED. For all cases, LiMU approaches LMM with a higher value of M. For a small value of M, LiMU performs slightly better than LMM when there is only a single significant locus, and LMM performs better than LiMU when there are multiple significant loci.

AUC as a function of M provided by LiMU, LM, LMM, and the FIXED in four scenarios with number of significant loci d = 1 (upper left), 2 (upper right), 5 (lower left), and 10 (lower right).
We carried out a GWAS using the LiMU method to control for population structure for 467 TB subjects (of which 158 had MDR strains) collected in Lima, Peru. These data were previously studied in Grandjean et al (2017), and in that study, many homoplastic variant sites were identified to be significantly correlated, indicating epistasis. Our analysis further refines these results with the LiMU control for population structure. We removed genotypes with minor allele frequency <
We compared LM, the fixed effects model with the genetic similarity matrix represented by MDS implemented in software pyseer, LMM, and the LiMU. For the LMM, we use the empirical genetic similarity matrix, and the inference was carried out by the EMMA method. For LiMU, we first ran MrBayes to get posterior samples of trees. We ran MrBayes with 1 million iterations, with burn-in given by the first half of the chain, and we collected 50 thinned posterior tree samples. Given the expected genetic similarity matrix based on each sampled tree, we use the EMMA method to infer the LMM parameters.
The genetic similarity matrix of the fixed effects model in pyseer was computed using the consensus tree provided by the MrBayes analysis. We consider MDR as the phenotype of interest, and form a binary variable indicating MDR or non-MDR. All samples identified as resistant to either rifampicin or isoniazid (but not resistant to other drugs) are included in the non-MDR set.
We compared our methods with a classical linear regression GWAS with t-tests. This linear analysis identified 100 genetic variants that significantly associated with MDR after Bonferroni (BF) correction, with p value

The base pair positions of LiMU and LMM hits. BP, base pair.

Manhattan plot of genome-wide association studies carried out by LM, the FIXED, linear mixed model (LMM), and LiMU for the TB data. The dashed horizontal line indicates the threshold after BF correction. The base pair positions of LiMU hits are also provided. BF, Bonferroni; TB, tuberculosis.
The fixed effects model implemented in pyseer software identifies 96 associated genetic variants (gray crosses) after BF correction. Both LMM and LiMU significantly correct hits found through linear regression, suggesting that many of these hits are due to population structure. Figure 8 displays the Manhattan plot for these GWASs. Table 2 gives the p values of the three most significant hits identified by LiMU. We also summarize the posterior p values through posterior median and geometric mean, yielding values that are close to the p values summarized by mean (the
Negative Log p Values of LM, LMM, LiMU, and pyseer for Base Pair Positions
LM, linear model; LMM, linear mixed model; LiMU, linear mixed model with uncertain genetic similarity matrix; BP, base pair.
The second row of Table 3 reports the timing (in seconds) for the three main steps of LiMU (i.e., 1 million iterations of MrBayes, computation of the genetic similarity matrix, and REML for one LMM fit). One million iterations of MrBayes run costs
Timing (in Seconds) of Each Step in LiMU for Simulation 2 (Row 1) and Real Analysis (Row 2)
The experiments were conducted on a 2.3 GHz Intel Core i9 processor.
LiMU, linear mixed model with uncertain genetic similarity matrix; GSM, genetic similarity matrix; REML, restricted maximum likelihood.
The hits with BP positions 761,160 and 761,115 are nonsynonymous mutations (these alter the amino acid produced) in the rpoB gene, which is associated with rifampin resistance (Goldstein, 2014; Lipin et al, 2007). The majority of mutations that confer resistance to rifampin occur within an 81 bp region of rpoB, referred to as the rifampin-resistance determining region (RRDR). And although neither of the sites identified here occur within the RRDR, there is still a chance that strains carrying these mutations may be rifampin resistant, or they may be compensatory mutations (Lempens et al, 2018; Ma et al, 2021).
In addition, the hit with BP position 2,155,176 is a nonsynonymous mutation in the katG gene, which is associated with isoniazid resistance. Rifampin and isoniazid are first-line antimicrobials used to treat TB and strains resistant to both are termed MDR-TB (Lipin et al, 2007).
There was also a hit identified by LiMU for a nonsynonymous mutation within rpoC, which has been previously shown be involved in compensation of fitness costs associated with rifampin resistance (De Vos et al, 2013). In addition, there were hits within various Pro-Pro-Glu (PPE) and Pro-Glu and polymorphic guanine-cytosine-rich repetitive sequence (PE-PGRS) family genes, and although the exact function of many of these genes is not well understood, there is evidence that many are involved in the host–pathogen interaction and infection (Qian et al, 2020). However, there can be technical challenges with assembly and variant calling at these loci because of a high GC content and excess of repetitive sequences (Ates, 2020), and further study would be required to validate the variation found in these genes.
Standard linear mixed models (LMMs) for GWASs often assume a single known genetic similarity matrix as a random effect (typically computed as the symmetric matrix resulting from inner products of genetic variants). However, such an approach is inaccurate if genotypes are not densely sampled, or are of poor quality (Wang et al, 2021): in Wang et al (2021), it was found that uncertainty in genetic similarity matrices (measured in standard deviation) varied from
We have developed a linear mixed effects model for GWASs incorporating uncertainty about the genetic similarity matrices, in which the genetic similarity matrix is induced by a phylogeny based on the genotype. To account for the uncertainty of phylogeny, we considered a Bayesian framework for the underlying tree and derive the posterior samples through MCMC methods (i.e., MrBayes). Our proposed method, LiMU, is computationally more expensive than standard LMMs as we require multiple runs of standard LMM, and use Bayesian sampling methods to obtain posterior tree samples. However, LiMU allows us to consider the uncertainty in the genetic similarity matrix (or phylogeny).
In LiMU, we first estimate posterior samples for the phylogeny, and then estimate parameters of the LMM conditioned on the trees. Our method can utilize any Bayesian phylogenetic inference methods that exist in current literature (Bouckaert et al, 2014; Ronquist et al, 2012; Wang and Wang, 2021; Wang et al, 2020). In addition, our method is flexible in the sense that the estimates of phylogenies could be obtained from DNA, RNA, or any data source arising from trees (including phylolinguistic data, for example).
Our simulations demonstrate the consistency of our methods, and improved FPRs over the LMM and the fixed effects model implemented in pyseer software. The ROC curve and AUC provided by LiMU dominate those provided by the LMM, the fixed effects model implemented in pyseer software, and a linear model. Our simulations further show that the advantage of LiMU is seen most clearly when the heritability of the phenotype is high. There is more uncertainty in the phylogeny for smaller data sets, and in this case LiMU is preferable.
We also demonstrate that LiMU is robust to model misspecification and high genotyping error (LiMU outperforms other methods in simulations with high genotyping error, and with simulations in which the phenotypes are not sampled from an LMM). We recommend that LiMU be used for data sets with high genotyping error or small number of markers. Our experiments involved <10,000 markers. If the number of markers is much larger, then the genetic similarity matrix will have less uncertainty and LiMU results may approach those of the LMM.
We apply our method to a GWAS of 467 MDR-TB (with ∼10,000 markers) in a population from Lima, Peru. In our real data analysis, LiMU found fewer hits than a linear model without random effects. The hits we found involve nonsynonymous mutations in the rpoB and katG genes, and a nonsynonymous mutation within rpoC, that is associated with rifampin resistance. These genes are known to be involved with MDR or host–pathogen interaction and infection. Our simulations suggest that the FPR of LiMU is lower than that of the LMM, and so these hits are likely to be true positives. Also, the hit we identified at BP position 2,155,176 was not found by the LMM.
Our current approach is limited to sequences without recombination. We could extend to data with recombination events in genealogies. The ancestral recombination graph (ARG) describes the coalescence and recombination events among individuals (Rasmussen et al, 2014). The ARG is composed of a set of coalescent trees separated by break points. To compute expected genetic similarity matrix for samples given an ARG, we could first compute the expected genetic similarity matrices for each of the coalescent trees in the ARG, then compute weighted average for those expected genetic similarity matrices.
The weights would be proportional to the number of loci between each consecutive pair of break points. Finally, we could apply LiMU to this weighted set of trees, computing their expected genetic similarity matrices. In cases wherein reconstructing ARG is computationally too intensive (e.g., whole genome sequences), we could use methods such as Fastgear (Mostowy et al, 2017), Clonalframeml (Didelot and Wilson, 2015), and Gubbins (Croucher et al, 2015) to remove recombination from alignments before phylogenetic reconstruction.
Footnotes
AUTHORs' CONTRIBUTIONS
S.W. and S.G. contributed to methodology, formal analysis, software, writing—original draft, and writing—review and editing. B.S. contributed to writing—original draft and writing—review and editing. L.G. contributed to data curation. L.W. contributed to writing—review and editing. C.C. contributed to conceptualization and writing—review and editing. L.T.E. contributed to conceptualization, methodology, writing—original draft and writing—review and editing.
ACKNOWLEDGMENTS
We thank the associate editor and anonymous referees who provided helpful comments to this article.
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
This research is supported by the National Natural Science Funds of China (Grant No. 12101333), the Natural Science Funds of Tianjin (Grant No. 21JCQNJC00050), the Shanghai Science and Technology Program (Grant No. 21010502500), the startup fund of ShanghaiTech University, and NSERC (Grant Nos. RGPIN/05484-2019, RGPIN/2019-06131, and DGECR/00118-2019). L.T.E. is supported by a Michael Smith Health Research BC Scholor Award.