
Abstract
The scientific community is rapidly generating protein sequence information, but only a fraction of these proteins can be experimentally characterized. While promising deep learning approaches for protein prediction tasks have emerged, they have computational limitations or are designed to solve a specific task. We present a Transformer neural network that pre-trains task-agnostic sequence representations. This model is fine-tuned to solve two different protein prediction tasks: protein family classification and protein interaction prediction. Our method is comparable to existing state-of-the-art approaches for protein family classification while being much more general than other architectures. Further, our method outperforms other approaches for protein interaction prediction for two out of three different scenarios that we generated. These results offer a promising framework for fine-tuning the pre-trained sequence representations for other protein prediction tasks.
INTRODUCTION
The advent of new protein sequencing technologies has accelerated the rate of protein discovery (Restrepo-Pérez et al., 2018). While protein sequence repositories are growing exponentially, existing methods for experimental characterization are not able to keep up with the present rate of novel sequence discovery (Oh et al., 2018; The UniProt Consortium, 2018). Currently, less than 1% of all amino acid sequences in the UniProtKB database have been experimentally characterized (The UniProt Consortium, 2018). The explosion of uncharacterized proteins presents opportunities in computational approaches for protein characterization.
Harnessing protein sequence data to identify functional characteristics is critical to understanding cellular functions and developing potential therapeutic applications (Chen et al., 2019). Sequence-based methods to computationally infer protein characteristics have been critical for inferring protein function and other characteristics (Smith et al., 1981). Thus, the development of computational methods to infer protein characteristics (which we generally describe as “protein prediction tasks”) has become paramount in the field of bioinformatics and computational biology. Here, we adapt a Transformer neural network to establish task-agnostic representations of protein sequences, and use the Transformer network to solve two protein prediction tasks.
Deep learning for natural language processing tasks
In applying deep learning to sequence-based protein characterization tasks, we first consider the field of natural language processing (NLP), which aims to analyze human language through computational techniques (Manning and Schütze, 1999). Deep learning has recently proven to be a critical tool for NLP, achieving state-of-the-art performance on benchmarks for named entity recognition, sentiment analysis, question answering, and text summarization, among others (Young et al., 2017).
Neural networks are functions that map one vector space to another. Thus, to use them for NLP tasks, we first need to represent words as real-valued vectors. Often referred to as word embeddings, these vector representations are typically “pre-trained” on an auxiliary task for which we have (or can automatically generate) a large amount of training data. The goal of this pre-training is to learn generically useful representations that encode deep semantic and syntactic information (Young et al., 2017). Then, these representations can be used to train systems for NLP tasks for which we have only a moderate amount of training data.
Word embeddings can be broadly categorized as either context-free or contextualized. Methods such as Skip-Gram (as popularized by the seminal software package word2vec) and GloVe generate context-free word vector representations (Mikolov et al., 2013; Pennington et al., 2014). Once trained, these methods assign the same embedding to a given word independent of its context. Contextualized embedding models such as OpenAI-GPT (Radford et al., 2019) and Embedding from Language Model (ELMo) (Peters et al., 2018) were later developed to generate on-demand contextualized embeddings given a word and its surrounding sequence. Contextualized word representations have been shown to have superior performance to context-free representations on a variety of benchmarks (Peters et al., 2018), and have become the new standard practice in NLP.
Building upon these methods, Devlin et al. (2019) developed a state-of-the-art contextualized word embedding technique called Bidirectional Encoder Representations from Transformers (BERT), to create “deeply bidirectional” embeddings using transformers. BERT's architecture includes multiple Transformer encoder layers (Vaswani et al., 2017), whose architecture we explain in Section 2.2, to construct a model that generates token representations by simultaneously incorporating the leftward and rightward context of sentences. Recently, a new procedure called Robustly Optimized BERT Pretraining Approach (RoBERTa) was developed that improved BERT's performance on the original masked language task (Liu et al., 2019).
From NLP to protein prediction tasks
Because of the unprecedented success in applying deep learning to NLP tasks, one of the recent interest areas in computational biology has been applying NLP-inspired techniques to amino acid sequence characterization. These techniques typically treat amino acids as analogous to individual characters in a language alphabet. Following the pre-training regime established in NLP, tools such as seq2vec (Heinzinger et al., 2019) and ProtVec (Asgari and Mofrad, 2015) have been developed to create amino acid sequence embeddings for protein prediction tasks. These two representation methods are based on the ELMo and Skip-Gram technique, respectively, and demonstrate state-of-the-art accuracy when applied to bioinformatics tasks.
Inspired by the success of BERT in NLP, in this article we pre-train a Transformer network on amino acid sequence representations for protein prediction tasks. Our model first appeared at a conference in 2020 (Nambiar et al., 2020), and similar Transformer neural networks have been proposed for protein prediction tasks in recent years. In fact, Transformer neural networks have become the workhorse of the protein representation learning, with multiple publicly available pre-trained models such as ESM-1b (Rives et al., 2021) and ProtBert (Elnaggar et al., 2021). These models can be distinguished based on several characteristics including the size of the neural network, the dataset it was pre-trained on, and how sequences were tokenized (Table 1).
Characteristics of Three Different Bidirectional Encoder Representations from Transformers-Inspired Protein Language Models
Characteristics of Three Different Bidirectional Encoder Representations from Transformers-Inspired Protein Language Models
PRoBERTa is the model used in this work.
BFD, Big Fat Database.
The largest model, ESM-1b, was trained on the UniRef50 dataset where 250 million sequences were clustered at 50% sequence identity and a representative sequence from each cluster was used (Rives et al., 2021). Using UniRef50 could be advantageous because the clustering of sequences decreases the redundancy of sequences seen by the model. The disadvantage of using UniRef50 is that this dataset might not allow the model to learn evolutionary information as easily, which has been addressed in other work by clustering using 90% sequence identity (Meier et al., 2021). ProtBert, on the other hand was trained on the Big Fat Database (BFD) that is made up of over 2 billion proteins. This increase in number of proteins was due to the inclusion of metagenomic data in the dataset, allowing the model to be trained on a wider range of sequences (Elnaggar et al., 2021).
However, the fact that a large fraction of the dataset is made up of sequences obtained from metagenomic data could also be a source of bias in the data. Finally, we use the SwissProt dataset that contains only 450,000 reviewed sequences for Protein RoBERTa (PRoBERTa) (Nambiar et al., 2020). The benefit of using SwissProt is that since all of the sequences are reviewed it is less likely that the dataset contains low quality sequences. This was especially important for us since one of the goals of our work was to pre-train a model with a small number of sequences, to be cost efficient.
This need for cost efficiency also led to our use of byte-pair encoding (BPE) for sequence tokenization, a characteristic that is not shared by other transformer-based protein language models. The differences between models allow researchers to use the model that best fits their downstream tasks and computational budget. These and similar models have been used for various tasks including protein localization prediction, variant effect prediction, and protein contact prediction, among others (Bhattacharya et al., 2020; Elnaggar et al., 2021; Meier et al., 2021; Stärk et al., 2021). In this article, we focus on using our pre-trained models for two tasks: protein family prediction and protein interaction prediction.
Protein families are groups of evolutionarily-related proteins that typically share similar sequence, structural, and functional characteristics. By taking advantage of the evolutionary conservation of amino acid sequence and structure, resources such as CATH-Gene3D, PANTHER, Pfam, and SUPERFAMILY cluster amino acid sequences that share an inferred origin into hierarchical groups known as protein superfamilies, families, and subfamilies (Gough et al., 2001; Punta et al., 2011; Mi et al., 2015; Dawson et al., 2017). Traditionally, family classification has required the comparison of experimentally identified characteristics. However, methods have also been developed to computationally classify proteins based solely on sequence similarity. This approach enables us to infer functional and structural characteristics of proteins in a high-throughput manner.
Current computational approaches for protein family classification include methods such as BLASTp and profile hidden Markov models (pHMMs) that compare sequences to a large database of pre-annotated sequences. However, inference using these alignment-based methods is computationally inefficient, as they require repeated comparison of sequences to an exponentially growing database of labeled family profiles and are limited by expensive, manually tuned processing pipelines (Das and Orengo, 2016). With the exponential growth of protein discovery, the development of more scalable approaches is required to overcome traditional bottlenecks (Bileschi et al., 2022).
Guided by a deep learning framework, recent models based on convolutional neural network (CNN) and recurrent neural network (RNN) architectures have been successful in achieving state-of-the-art accuracy on the protein family classification task (Heinzinger et al., 2019; Bileschi et al., 2022). However, these methods still produce task-specific models that are unable to generalize toward a broader range of protein prediction tasks. One recent model, UDSMProt, used an RNN architecture in a similar pre-training and fine-tuning framework to predict whether a given protein is contained in the same superfamily or fold of a reference protein (Strodthoff et al., 2020). Such an approach requires pairwise comparison of sequences against multiple reference proteins, which may not be entirely representative of a protein family.
Task: Protein–protein interaction prediction
The next protein prediction task we highlight is protein
Identifying PPIs and creating PPI networks, or interactome networks, has thus become central to the study of biological systems (De Las Rivas and Fontanillo, 2010). By mapping these relationships, we can understand the complex interactions between individual components in a living system with a holistic approach. For instance, through the comparative analysis of PPIs in both healthy and diseased states, we can study disease mechanisms as a whole and identify potential therapeutic targets (Kuzmanov and Emili, 2013; Petta et al., 2016). However, experimental identification of PPIs has proven to be a complex and time-consuming process, thus creating the need for an efficient and reliable method of computationally predicting PPIs.
Traditionally, computational identification of PPIs has relied on genomic, structural, or domain information of the interacting proteins (Guo et al., 2008). However, such knowledge is not readily available for most proteins. Instead, sequence-based identification currently relies on domain-based methods such as support vector machines and random forest classifiers that extract features such as amino acid distributions and domain compositions. These current approaches have limited information extraction capability and demonstrate low prediction accuracy (Chen and Liu, 2005; Zhang et al., 2014; Alonso-López et al., 2019). More recent work has leveraged deep learning-based architectures such as stacked autoencoders, RNNs, and recurrent convolutional neural networks (RCNNs) (Sun et al., 2017; Chen et al., 2019; Guo and Chen, 2019). These models have achieved state-of-the-art accuracy in the binary PPI classification task, and the ability to generalize to similar PPI characterization tasks such as interaction type prediction and binding affinity estimation (Chen et al., 2019; Guo and Chen, 2019).
Contributions
We apply a Transformer neural network, which we call PRoBERTa, to pre-train task-agnostic vector representations of amino acid sequences. PRoBERTa modifies the RoBERTa procedure by reducing the number of transformer layers, using the Layerwise Adaptive Moments optimizer for Batch training (LAMB), and training the model on amino acid sequences tokenized using BPE (You et al., 2019). We then fine-tune these representations toward two protein prediction tasks: protein family classification and PPI prediction.
We present two variants for the protein family classification task: (1) a multi-class classification problem of predicting a family label for a given sequence and (2) a binary classification problem of predicting whether a sequence is a member of a chosen family label. We show that the embeddings produced by PRoBERTa can be used to produce models for family classification that contain more information about protein family membership than the pre-trained embeddings, and have comparable performance to current methods that use specialized task-specific architectures. We define PPI prediction as a binary classification task to predict whether two proteins will interact given their amino acid sequences. We present three experimental settings for binary PPI prediction based on how negative examples (non-interacting proteins) are chosen. PRoBERTa outperforms other methods in two out of the three settings.
PRoBERTa is also much more computationally efficient than past work that applies Transformer networks to encode protein sequences to predict protein secondary structure. Using a BERT-based model, Rives et al. (2021) pre-trained their model on 128 NVIDIA V100 GPUs for 4 days. In comparison, we pre-train PRoBERTa on four NVIDIA V100 GPUs in 18 hours using (1) a modified architecture, (2) the RoBERTa training procedure (Liu et al., 2019), and (3) the LAMB optimizer (You et al., 2019). By using this framework, we can use a smaller pre-training corpus while obtaining state-of-the-art accuracies, increasing the computational efficiency for pre-training by a factor of 170 compared to the most recently published model.
METHODS
We treat proteins as a “language” and draw ideas from the state-of-the-art techniques in NLP to obtain a vector representation for proteins. For a sequence of amino acids to be treated as a sentence, the alphabet of the language is defined such that

Each symbol
Tokenization with BPE
Before amino acid sequences can be interpreted as a language, we must first define what a word is. This is more challenging for proteins than most natural languages because unlike the space character in languages like English, there is no single character (or amino acid) that is used to divide parts of an amino acid sequence into meaningful chunks. In the past, deep learning models have either used individual amino acids as input (Hashemifar et al., 2018; Chen et al., 2019) or have chosen to group every three amino acids as a “word” (Asgari and Mofrad, 2015). However, there has been recent interest (Asgari et al., 2019) in statistically determining segments of amino acids to be used as inputs for downstream machine learning algorithms using a method called BPE (Gage, 1994). BPE was originally developed as a compression algorithm although it has been adapted more recently as an NLP method for identifying subword units (Sennrich et al., 2016).
In our application, given an amino acid sequence
Transformer network architecture
Inspired by the BERT language representation model architecture, PRoBERTa consists of (1) an embedding layer, followed by (2)
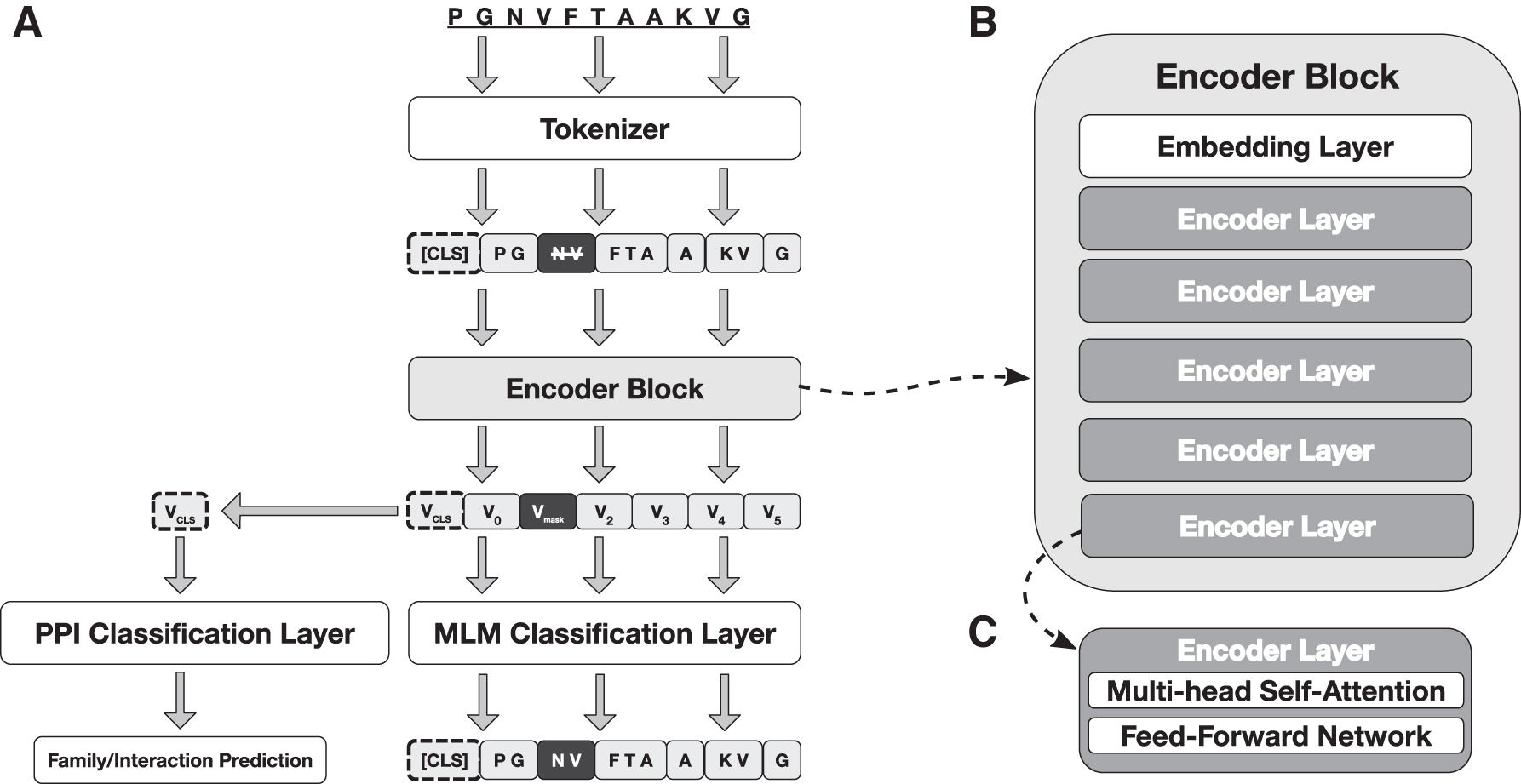
PRoBERTa architecture for pre-training and fine-tuning.
A tokenized amino acid sequence
where
Embedding layer
To prepare an input sequence
Transformer encoder layer
Each Transformer encoder layer contains two sub-layers—a multi-head self-attention mechanism and a fully-connected feed-forward network—with residual connections around each sub-layer followed by a layer normalization operation (Ba et al., 2016). Each sub-layer, and thus the entire encoder layer, takes as input and produces a list of n vectors, each of dimension
Given an input list of vectors
The initial input vector xi is then added to the output vector yi, after which yi undergoes a layer-normalization step and passes through a fully connected feed-forward network that has a single hidden layer of size
Model output
Without adding any task-specific heads to the architecture, the model output is a list of
Model pre-training
Following the BERT framework, we train PRoBERTa in two stages: pre-training and fine-tuning (Devlin et al., 2019). In the pre-training stage, our objective is to train the model to learn task-agnostic deep representations that capture the high-level structure of amino acid sequences.
Based on the RoBERTa procedure, we pre-train PRoBERTa using only the unsupervised masked language modeling (MLM) task, which adds a Language Modeling head to the network architecture. MLM randomly masks certain tokens and then trains the network to predict their original value (Liu et al., 2019). We expect MLM to be a useful task because it will train the neural network to predict groups of amino acids in a sequence based on the other amino acids and their order in the sequence. This should impart some general-purpose, biologically relevant information regarding the sequences to the network. Specific training hyperparameters and optimization are detailed in Section 2.5.
Given a tokenized input sequence
Pre-training data
We use UniProtKB/Swiss-Prot (450K unique sequences with a mean tokenized length of 129.6 tokens), a collection of experimentally reviewed amino acid sequences (The UniProt Consortium, 2018). Sequences are tokenized with the BPE algorithm described in Section 2.1. In our experiments, the maximum vocabulary size was set to 10,000 because we empirically observed that the mean token length increased very little beyond 10,000 tokens, indicating that most of the longer tokens were detected in the first 10,000 iterations.
Model fine-tuning
The pre-trained model can then be specialized for downstream protein prediction tasks. In the fine-tuning stage, we initialize the model with the pre-trained parameters. We then modify the pre-trained architecture by replacing the output layer with a task-specific layer with dimensions tailored to the specific task. Parameters are fine-tuned using labeled data from the prediction tasks. Here, we fine-tune the pre-trained model for our two specific prediction tasks: family classification and PPI prediction. For our selected tasks, we feed the aggregate sequence representation corresponding to the special
Task: Protein family classification
For this task, we perform two modes of classification: binary family classification and multi-class family classification. In binary family classification, we train a separate classifier for each protein family to identify which sequences belongs to a given family. This classifier performs logistic regression on the trained sequence representations from the pre-trained model. We create a balanced training dataset for each classifier consisting of all the positive examples and the same number of negative examples drawn uniformly at random without replacement from outside the family. In multi-class family classification, we train a single classifier that outputs a probability distribution over the set of all protein families. In both classification modes, we require amino acid sequences to have membership in only one protein family for ease of classification.
Fine-tuning data
For the Family Classification tasks, we use 313,214 unique protein sequences from UniProtKB/Swiss-Prot whose manually curated annotations include protein family information and are not associated with multiple families or hierarchical family classifications (The UniProt Consortium, 2018).
Task: PPI prediction
Given a pair of tokenized amino acid sequences
we pack them together into a single input sequence separated by a special token
We truncate each tokenized amino acid sequence to 254 tokens before concatenation so the maximum combined length of the input sequence after the addition of the special tokens is
2.4.2. 1. Fine-tuning data
For the PPI prediction task, we use experimentally identified human PPIs from the Human Integrated Protein-Protein Interaction rEference (HIPPIE) database that are confidence scored and functionally annotated (Alanis-Lobato et al., 2016). Because HIPPIE only reports interacting protein pairs, we generated three sets of putative non-interacting protein pairs to serve as negative examples. In the “conservative” scenario, we generated 275,401 pairs using randomly selected human proteins from UniProt (Hamp and Rost, 2015) that are not reported to interact in HIPPIE, resulting in a PPI dataset of 536,545 pairs. This random generation of negative examples is possible because of the assumption that PPI networks are sparse, although we note the possibility that these negative examples may include interacting protein pairs not reported in HIPPIE.
In the “aggressive” scenario, we generated 275,401 pairs using randomly selected proteins from HIPPIE that are not reported to interact with each other, also resulting in a PPI dataset of 536,545 pairs. Finally, in the “ultra-aggressive” scenario, the negative examples are generated by performing a weighted sampling of pairs of proteins from HIPPIE that do not interact with each other. Here the weights used are the average degree of the proteins in the HIPPIE network. Therefore, the ultra-aggressive negative space will have pairs of proteins that are highly likely to interact with other proteins but not with each other (Fig. 2). However, it is important to note that the ultra-aggressive scenario is most likely to include negative examples that may be positives that have not been experimentally tested.
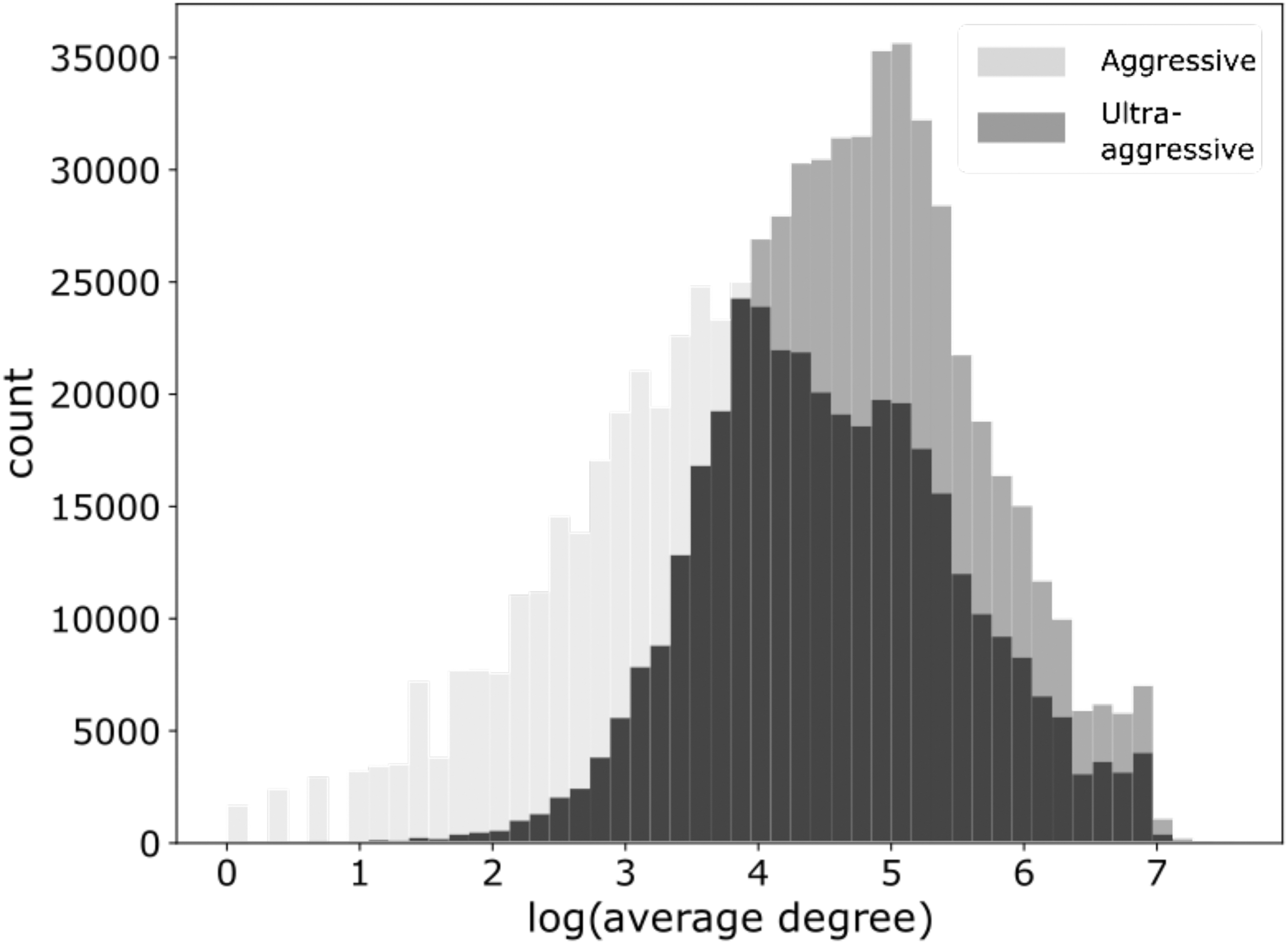
Average degree of negative interacting proteins for the aggressive and ultra-aggressive scenarios. The average degree of the negative protein pairs are shown on the x-axis. Negative pairs in the ultra-aggressive scenario are chosen by weighted sampling based on these average degrees.
We use the
Model Hyperparameters for Training
Model Hyperparameters for Training
We train our model on the cross-entropy loss with the LAMB optimizer (You et al., 2019), which is a layerwise adaptive large batch optimization technique developed to increase performance and reduce training time for attention-based models. The learning rate is warmed up linearly over to the peak value. Afterward, it is adjusted using a polynomial decay policy. For pre-training, we selected the learning rate and warmup period using the square root learning rate (LR) scaling heuristic and linear-epoch warmup scheduling because of their success when applied to BERT-based models (You et al., 2019).
To avoid overfitting and balance model performance with computational efficiency, we use early stopping with a patience value of 3 (training stops after 3 consecutive epochs with no improvement in either MLM validation loss during pre-training or task-specific validation accuracy during fine-tuning).
For both protein prediction tasks, we use the following metrics to evaluate a model's prediction. Accuracy is the proportion of predictions made by the model that are correct. Precision is the proportion of positive predictions made by the model that are correct. Recall is the proportion of correct positives that are identified by the model. Finally, the adjusted mutual information (AMI) is an entropy-based metric that measures the agreement between two sets of labels (Pedregosa et al., 2011). AMI adjusts mutual information to account for chance: For two labels U, V, we compute the AMI as
where
RESULTS
We first describe the sequence features learned from the pre-trained model. We then show PRoBERTa's performance when the model is fine-tuned for the Protein Family Classification and PPI Prediction tasks. Finally, we perform a robustness analysis by limiting the amount of labeled input data during fine-tuning.
Protein embeddings from the pre-trained model
We pre-trained the PRoBERTa model as described in Section 2.3 on 4 NVIDIA V100 GPUs in 18 hours. We first explored whether the pre-trained model captured any biological meaning from the amino acid sequences. We created protein embeddings by concatenating the vectors of each protein's first 128 tokens. We concatenated the vectors because the concatenated vectors appeared to provide better visualization results than the just the [CLS] (described in Section 2.2.1) token. The pre-trained model is already able to distinguish between protein families; Figure 3 shows the first two principal components of thirty randomly selected proteins from seven related protein families.

The first two principal components of pre-trained embeddings for 189 amino acid sequences from seven representative protein families.
To systematically evaluate how well the pre-trained protein embeddings distinguish protein families, we clustered 9151 protein embeddings from the manually annotated human proteins in UniProt that belong in families with more than one protein and compared the clusters to the 1761 annotated protein families using AMI. To cluster embeddings, we summed the representations for each token and reduced these embeddings to twenty dimensions using PCA and applied k-means clustering using Euclidean distance, setting k = 1800 to approximate the number of annotated families. The mean AMI of the k-means clusters, averaged over 20 runs, is 0.328, which is significantly higher than the expected AMI of 0 for randomly assigned clusters (Fig. 4).
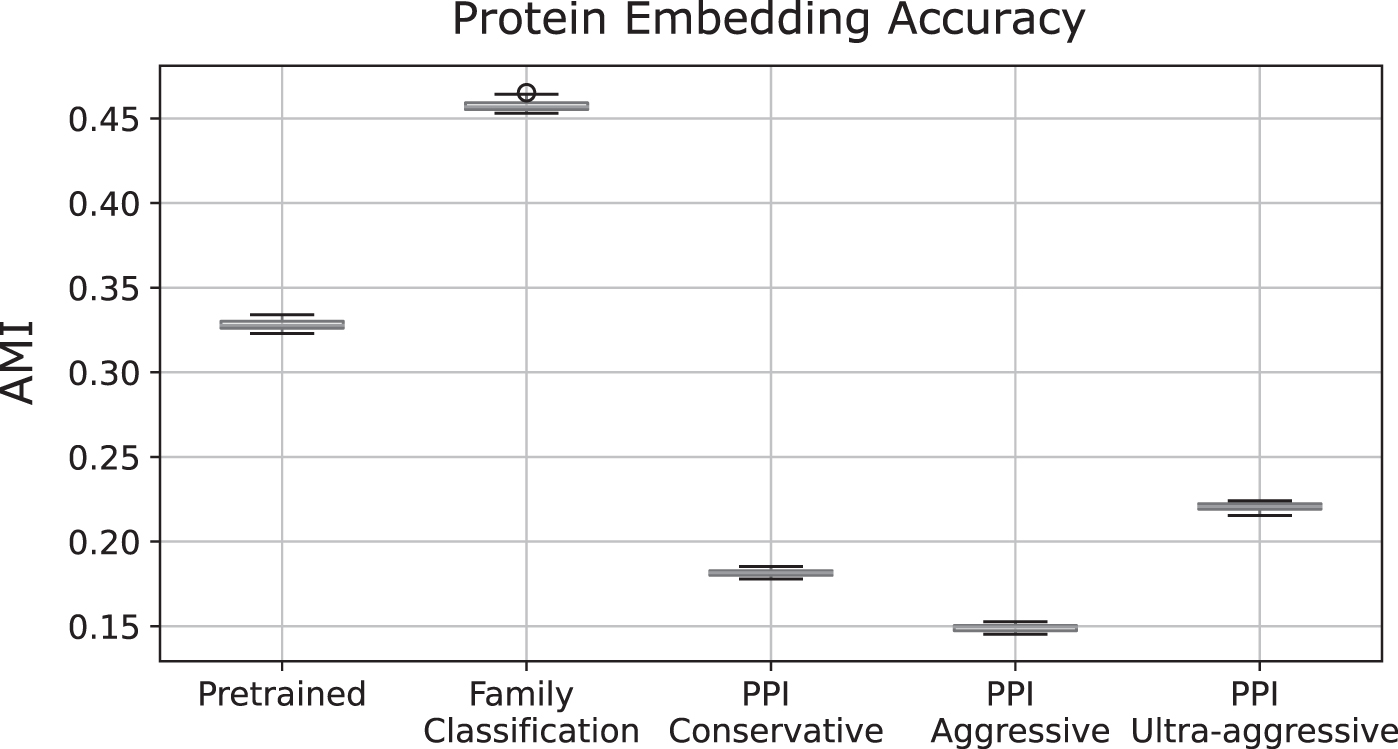
Accuracy of unsupervised clustering of three different versions of PRoBERTa embeddings with the true protein families given by UniProt. AMI values are shown for the pretrained embeddings and the fine-tuned embeddings for the protein prediction tasks. AMI, adjusted mutual information.
Given the promise of the protein embeddings, we then evaluated the performance of the PRoBERTa model on the protein family classification task (Section 2.4.1). Clustering the embeddings after fine-tuning on the protein family classification task shows a higher AMI than clustering after pre-training (Fig. 4), suggesting that the fine-tuned embeddings capture more protein family information.
For the binary classification task, we trained a separate logistic regression classifier for each protein family with more than 50 proteins and measured the weighted mean accuracy as 0.98. The classifier corresponding to the lowest scoring family, made up of 57 proteins, had an accuracy of 0.77. To train these classifiers, we randomly withheld 30% of the proteins from each family to be used as the test set. We compared PRoBERTa+logistic to three other NLP based embedding methods: ProtVec, which is a protein embedding method inspired by Word2Vec; ProtDocVec, which modifies ProtVec to use Doc2Vec; and ProtFreqVec, which uses the frequency of triplets of amino acids to form embedding vectors (Asgari and Mofrad, 2015). PRoBERTa+logistic performs better than ProtVec+logistic and similarly to ProtDocVec+logistic and ProtFreqVec+logistic (Table 3).
Comparison of Binary (Left) and Multi-Class (Right) Family Classification
Comparison of Binary (Left) and Multi-Class (Right) Family Classification
CNN, convolutional neural network.
This result supports the idea that contextual embeddings do not significantly help binary family classification. In Figure 3a, all the families but the importin beta family were chosen from families that have accuracies that are representative of the overall accuracy (ranging from 0.96 to 1.0) while the importin beta protein family has a substantially lower accuracy of 0.77. The PCA plots in Figure 3 show that the proteins in this less accurately classified family are co-located with proteins from the FormylGlycinAMidine ribonucleotide Synthetase (FGAMS) family and transfer ribonucleic acid (tRNA) pseudouridine synthase TruA family.
In the multi-class family classification task, we used fine-tuning to add an output layer that maps to protein family labels to the PRoBERTa model. This was done using the dataset of 313,214 UniProt proteins with only one associated family. These proteins were split into train/validation/test sets (0.8/0.1/0.1), and our fine-tuned classifier achieved an accuracy of 0.93 on the test set. We then compared this to two other multi-class family classifiers including a simple CNN made up of four convolution layers as a baseline and DeepFam, a CNN method that is the current state-of-the-art method for protein family classification. In particular, DeepFam is made up of a convolution layer with eight different kernel sizes and 250 convolution units for each kernel size (Oh et al., 2018). PRoBERTa with a classification layer performed better than the baseline method and had comparable accuracy to DeepFam (Table 3).
We next assessed PRoBERTa on the PPI task using the conservative, aggressive, and ultra-aggressive scenarios for sampling non-interacting protein pairs (Section 2.4.2).
Conservative scenario
We first evaluated how well the fine-tuned model's embeddings capture protein family information compared to the pre-trained embeddings. In the conservative scenario, the fine-tuned model produces embeddings that cluster with a lower AMI with protein families compared to the pre-trained embeddings (Fig. 4), indicating that the parameters of the model fine-tuned on predicting interactions are not as tuned to protein family classification. Evaluated on the test set, the fine-tuned PRoBERTa PPI classifier had an accuracy of 0.95 with a precision and recall of 0.95 and 0.95, respectively (Table 4) and a receiver operating characteristic (ROC) area under the curve (AUC) of 0.99 when ranking the predictions by the softmax probability (Fig. 5A). In these runs, we only used 20% of the available training and validation data from the train/validation/test (0.8/0.1/0.1) split. This was done because some of the methods we compare against were not able to scale up to using 80% of the data for training and thus would not be able to make a fair comparison to the PRoBERTa model trained with the entire train set.

PPI Prediction Results with 20% of Training Data (Top) and 100% of Training Data (Bottom)
Bold = best performing method according to each metric.
DI, deep interact.
We compare our results to PIPR, which is one of the top PPI prediction neural networks currently available, using a similar number of interactions from our dataset (Chen et al., 2019). PIPR uses a residual convolutional neural network (RCNN) architecture to extract both sequential information and local features relevant for PPI prediction. We also compare our embeddings to the ProtVec embeddings combined with a feed-forward neural network with three hidden layers (which we call DeepInteract) that predicts PPI.
Finally, we try a biologically motivated transfer learning approach by first training a DeepFam network on protein family classification and then using one of the hidden layers as the vector representations of the proteins to be used by DeepInteract. As seen in Figure 5A, PRoBERTa with a classification layer outperforms all of these methods by a large margin. Further, when using the complete dataset, the accuracy reaches 0.98 (Table 4 and Fig. 6).
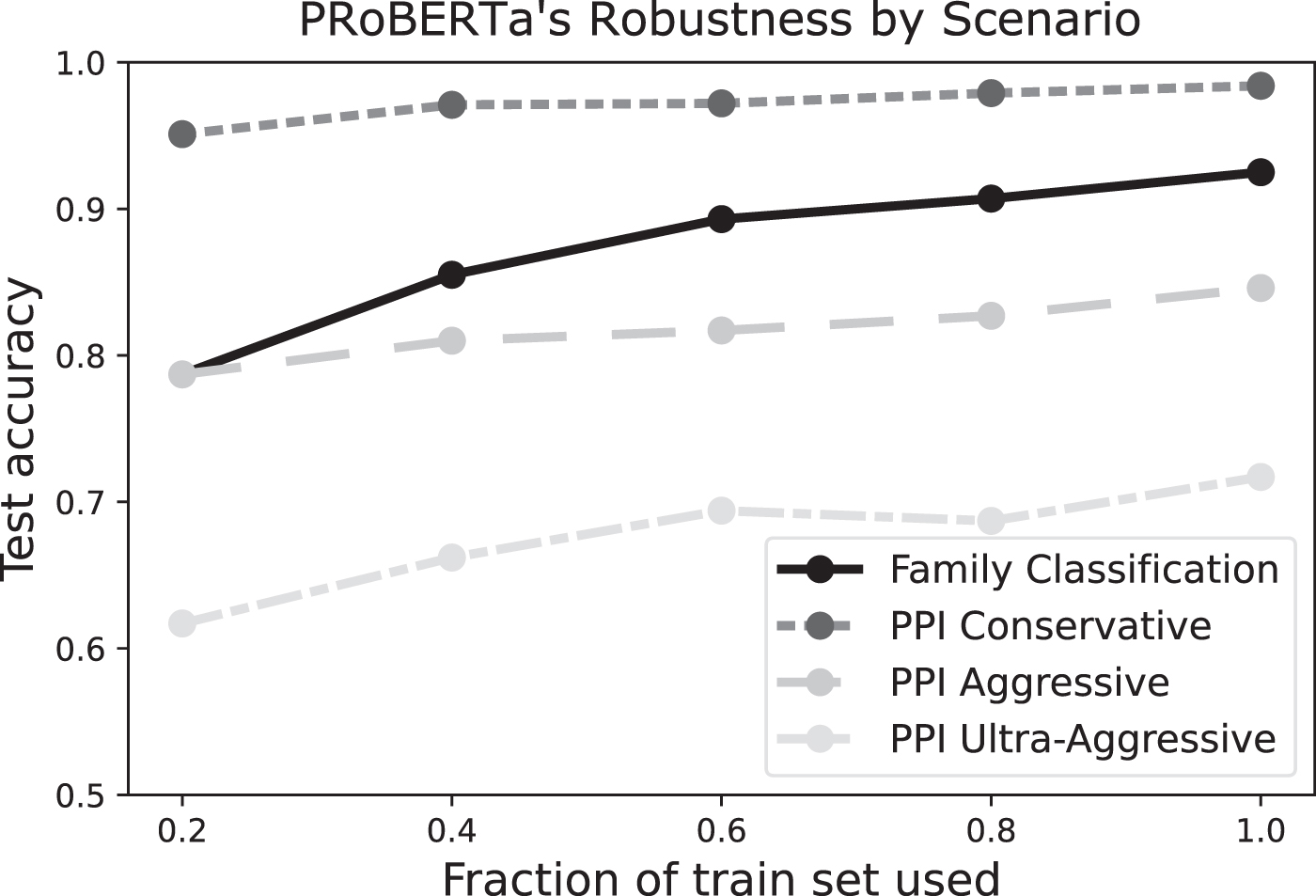
Accuracy on test data when using varying amounts of training data for fine-tuning on pre-trained models.
Similar to the conservative scenario, the model fine-tuned on the aggressive PPI dataset produces embeddings with a lower AMI with protein families than the pre-trained embeddings (Fig. 4). For this scenario, PRoBERTa had an accuracy of 0.79 with a precision and recall of 0.86 and 0.68, respectively (Table 4) and a ROC AUC of 0.88 (Fig. 5B). Similar to above, we only use 20% of the available training and validation data. As seen in Figure 5B, PRoBERTa with a classification layer still performs better than PIPR, which performs better than the other two methods. Moreover, when the full training dataset is used, the accuracy of our model improves to 0.85 (Table 4 and Fig. 6).
Ultra-aggressive scenario
Finally, in the ultra-aggressive scenario, the ultra-aggressive PPI model shows a lower AMI with protein families than the pre-trained embeddings, as expected based on the previous two PPI models (Fig. 4). For the ultra-aggressive dataset, PRoBERTa has an accuracy of 0.62 with a precision of 0.59, a recall of 0.71 and a ROC AUC of 0.67, showing that even in this challenging scenario, PRoBERTa is still able to make non-random predictions. However, as seen in Table 4 and Figure 5C, PRoBERTa is no longer the best performing method, with the fine-tuned DeepFam model obtaining a slightly higher accuracy of 0.63.
Pretraining improves performance in PPI prediction
To test how the pre-training step of PRoBERTa helps to improve its predictive power, we also trained Transformer models to predict PPI in all three scenarios without any pre-training. These models are otherwise identical to PRoBERTa. As shown on Figure 5D, pre-training improves the prediction accuracy in all three scenarios. This is another indication that the MLM task extracts biologically relevant information from unannotated protein sequences.
PRoBERTa scalability and robustness
We also investigated the robustness of the models by varying the amount of training data for both fine-tuning tasks. For the PPI prediction task in Section 3.3, we used 20% of the training data to compare to existing methods; here, we can use all of the training data, improving the performance in both the conservative and aggressive scenarios (Table 4).
To assess the robustness of PRoBERTa on the protein prediction tasks, we used fractions of the 0.8 and 0.1 split that made up the fine-tuning train and validation set, respectively, for task training. For example, if 90% of the train and validation set was used, this meant that 90% × 0.9 or 81% of the entire dataset was seen during training. Figure 6 shows the change in accuracy with different fractions of the train set used. This shows that all three models were somewhat robust to different amounts of training data. The PPI models appear to be more robust (they have smaller slopes) than the Protein Family model. However, it should be noted that the complete dataset for the Protein Family model contained 313,214 proteins, while the PPI dataset had 536,545 interactions in all three scenarios. The difference in robustness could be due to the absolute difference in the number of training data points.
DISCUSSION
In this article, we propose a Transformer based neural network architecture, called PRoBERTa, for protein characterization tasks. This neural network is based on the BERT architecture and the RoBERTa training procedure with a reduced number of Transformer layers and using the LAMB optimizer. We found that the pre-trained embeddings contain general yet biologically relevant information regarding the proteins and fine-tuning pushes the embeddings to have more specific information at the cost of generality. While there are notable differences between protein sequences and natural language corpuses (Luo et al., 2020; Strodthoff et al., 2020), leveraging the architecture from BERT tuning can capture this biologically relevant information. Altering the architecture to include prior knowledge unique to biological sequences could further improve the embedding space.
We found that using the embeddings for Protein Family Classification produced results that were comparable to the current best methods. In particular, we performed two different forms of classification; a binary classification that classified a protein as “in the family” or “not in the family” and a multi-class family classification. The multi-class family classification was based on the simplification that there is only one class per protein. Proteins that belong to more than one family were excluded from this classifier but not the binary classifiers.
Furthermore, we used embeddings from PRoBERTa for a fundamentally different problem, PPI prediction, using three different datasets generated from the HIPPIE database and found that with sufficient data, it substantially outperforms the current state-of-the-art method in the conservative scenario and still performs better than the other methods in the aggressive scenario. When evaluated on the aggressive dataset, the model trained on the conservative dataset scores an overall accuracy of 0.55, with a precision and recall of 0.52 and 0.99, respectively. This suggests that the model in the conservative scenario performs something closer to a protein classification task to identify which proteins are present in HIPPIE and are thus more likely to correspond to positive examples. The further drop in accuracy with the ultra-aggressive scenario may indicate that PRoBERTa's predictive power in the other scenarios is in part due to correctly predicting proteins that interact with many other proteins. Therefore, we are currently working on adapting PRoBERTa to predict protein promiscuity.
A concern that one might have in evaluating a model on these tasks is that there may have been very similar sequences in the train and test sets. To verify that this was not the case, we clustered the proteins in the HIPPIE dataset using Cluster Database at High Identity with Tolerance (CD-HIT) at a 90% sequence similarity threshold and found that very few proteins in the dataset were similar to each other (Huang et al., 2010). In particular, only 643 out of 16,215 proteins had sequence similarity of greater than 90% to another protein in the dataset and the average size of a cluster was 1.025.
The efficiency of PRoBERTa over existing methods [a speedup in pre-training time by a factor of 170 compared to the similar BERT-based model by Rives et al. (2021)] provides unprecedented opportunities for using the growing amount of sequence data in protein prediction tasks. Further, PRoBERTa's success in these two different protein prediction tasks alludes to the generality of the embeddings and the potential of smaller and more cost efficient transformer neural networks for a variety of protein prediction tasks. Finally, our use of BPE for sequence tokenization makes PRoBERTa particularly suitable for downstream tasks that involve long sequences.
AVAILABILITY
Scripts for pre-training, fine-tuning, and evaluating models, along with links to datasets and trained weights can be found at: https://github.com/annambiar/PRoBERTa
Footnotes
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
This work has been supported by the National Science Foundation (Award Nos. 1750981 and 1725729). This work has also been partially supported by the Google Cloud Platform research credits program (to A.R., M.H., and A.N.). S.L. has been supported by the James Scholar Honors Program and the Illinois Scholars Undergraduate Research Program. A.N. would like to thank Mark Bedau, Norman Packard, and the Reed College Artificial Life Lab for insightful discussions and Desiree Odgers for inspiring the idea of taking a linguistic approach to a biological problem.