
Abstract
Ordinary differential equations (ODEs) are widely used for elucidating dynamic processes in various fields. One of the applications of ODEs is to describe dynamics of gene regulatory networks (GRNs), which is a critical step in understanding disease mechanisms. However, estimation of ODE models for GRNs is challenging because of inflexibility of the model and noisy data with complex error structures such as heteroscedasticity, correlations between genes, and time dependency. In addition, either a likelihood or Bayesian approach is commonly used for estimation of ODE models, but both approaches have benefits and drawbacks in their own right. Data cloning is a maximum likelihood (ML) estimation method through the Bayesian framework. Since it works in the Bayesian framework, it is free from local optimum problems that are common drawbacks of ML methods. Also, its inference is invariant for the selection of prior distributions, which is a major issue in Bayesian methods. This study proposes an estimation method of ODE models for GRNs through data cloning. The proposed method is demonstrated through simulation and it is applied to real gene expression time-course data.
1. INTRODUCTION
Ordinary differential equation (ODE)
ODEs for GRNs provide information about the change in gene expressions, and they are constructed using functional relationships of genes identified by the GRNs and trajectories observed from time-course gene expression data (Bachmann et al, 2012; Kim, 2016). Once ODEs for GRNs are specified, ODE parameters should be estimated with observed time-course gene expression data. However, the estimation of ODE parameters is an important but challenging problem because of inflexible model nature of ODEs and noisy data. Time-course gene expression data typically have complex error structures such as heteroscedasticity, correlations between genes, and time dependency because they are usually obtained from high-throughput experiments, which are performed on biological samples using RNA-sequencing or microarray technologies at discrete time points. Hence, these characteristics of the data exacerbate the difficulties of estimating ODE models for GRNs.
To estimate ODE models, either a likelihood or Bayesian approach is commonly used. When we simply focus on fitting ODE solution to observed trajectories, the least square method can be used (Bates and Watts, 1988; Hemker, 1972; Li et al, 2005; Seber and Wild, 1989). Under independent Gaussian errors with constant variance, the least square estimation is equivalent to the maximum likelihood (ML) estimation. Various likelihood estimation methods for ODEs have been developed so far. Himmelblau et al (1967) proposed a method to convert ODEs into a nonlinear system using numerical quadrature, and de Boor and Swartz (1973) introduced a method for approximating ODE solutions using piecewise polynomial functions.
Bock (1981) developed a method for applying numerical optimization to individual time interval segments, which is a partition of the entire time interval. Varah (1982) proposed a two-step estimation method using regression splines. Also, Liang and Wu (2008) used a similar idea to two-step nonparametric regression in the measurement error framework. Ramsay et al (2007) proposed a two-step smoothing method known as generalized profiling method, which jointly takes into account smoothing of data trajectories and estimation of ODE parameters.
However, the likelihood approaches require optimizations for the estimation of ODE models, and these tasks are not easy. In practice, efficient optimization algorithms are required and their convergence should be guaranteed. Also, since the likelihood function surface is sensitive to ODE parameters, they may fail to attain the global optimum.
In contrast, Bayesian approaches are free from the optimizations and local optimum problems because they use sampling techniques to obtain the posterior distributions of ODE parameters. Huang et al (2006) and Huang and Wu (2006) estimated the ODE parameters of a HIV dynamic system using a hierarchical Bayesian method based on numerical ODE solutions. Another Bayesian method is to utilize the Gaussian process, which can provide a distribution over both fitted trajectories and ODEs (see Calderhead et al, 2009; Chkrebtii et al, 2016; Wang and Barber, 2014). Bhaumik and Ghosal (2015) extended the two-step smoothing idea into Bayesian estimation, and Huang et al (2020) proposed a one-step generative Bayesian model that directly combines nonparametric regression functions and ODEs. However, for small size of data, the posterior inference could depend much on prior information and it might not yield reliable inference (Kim, 2016).
To estimate ODE models for GRNs, Cao and Zhao (2008) and Campbell and Chkrebtii (2013) employed the generalized profiling method proposed by Ramsay et al (2007) under the assumptions of independence and constant error variance. Kim and Kim (2018) proposed an ODE model considering the complex error structures of time-course gene expression data and they developed an iterative estimation algorithm based on the generalized profiling method. However, this algorithm necessitates solving optimization problems iteratively. Hence, it is not easy to find the global optimum. On the contrary, if we consider Bayesian approaches for ODE models for GRNs, prior information could have a significant impact on the inference because gene experiments typically yield small size of data.
To overcome the drawbacks of both likelihood and Bayesian methods in ODE models for GRNs, this study proposes an estimation method based on data cloning. Data cloning was introduced by Lele et al (2007) and it provides ML estimates (MLEs) and their standard errors (SEs) using the Bayesian framework. It is a full Bayesian model that uses proper prior distributions and the likelihood function for K copies (clones) of original data. It is particularly useful for likelihood inference of complex models such as state-space models, mixed effect models, and nonlinear dynamic models (Lele et al, 2010; Lele et al, 2007). Although data cloning is an ML method, it is free from local optimum problems because it does not require any optimizations. Also, its inference is completely invariant to the choice of prior distributions. Therefore, the method proposed in this study can provide more reliable and accurate inference for ODE models for GRNs.
We introduce some background of this study in Section 2 and we propose an estimation method for ODE models for GRNs based on data cloning in Section 3. The proposed estimation method is verified through a simulation study, and then it is applied to the inference of the ODE model describing a GRN for zebrafish retina cell.
2. PRELIMINARIES
2.1. ODE model for GRN
In general, GRN analyses identify connections between transcription factors and targets as well as describe the dynamics of the identified networks. For the latter, ODEs are often used and they are helpful for understanding the whole gene regulation process (e.g., de Jong, 2002; Endy and Brent, 2001; Hasty et al, 2001; Karlebach and Shamir, 2008). ODEs for GRNs describe change of gene expression levels using their derivatives as follows:
where 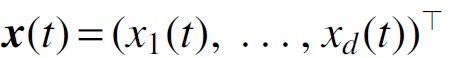 is a vector representing gene expression levels of d genes,
is a vector representing gene expression levels of d genes,
Time-course gene expression data are often collected from high-throughput gene experiments performed at discrete time points, and they quantify mRNA abundance for a huge number of genes at a time. Thus, time-course gene expression data typically have complex error structures. Subramaniam and Hsiao (2012) indicated that gene expression data from such experiments usually have heteroscedastic error depending on the expression level. In addition, since gene expression levels are measured for the same biological samples at each time point, the data have correlation between genes as well as their time dependency. ODEs are specified using a GRN and trajectories from time-course gene expression data.
Therefore, ODE models for such data should consider the complex but systematic error structures for more accurate prediction. To consider the complex error structures, Kim and Kim (2018) introduced a model as follows:
where 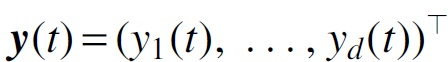 is a vector of expression levels of d genes observed at time t,
is a vector of expression levels of d genes observed at time t, 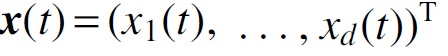 is a vector of ODE solutions from Equation (1),
is a vector of ODE solutions from Equation (1),
The variance function  is a parameter vector of the variance function for the ith gene. To illustrate correlation between genes and time dependency in the model, a vector autoregressive (VAR) model for the random vectors can be used. By assuming the Markov property, the VAR model with lag one is defined by
is a parameter vector of the variance function for the ith gene. To illustrate correlation between genes and time dependency in the model, a vector autoregressive (VAR) model for the random vectors can be used. By assuming the Markov property, the VAR model with lag one is defined by
2.2. Data cloning
Data cloning proposed by Lele et al (2007) is an ML estimation method using Bayesian MCMC, and it is particularly useful for estimating complex models such as hierarchical models, state-space models, mixed-effect models, and nonlinear dynamic models. Furthermore, this method is free from sensitivity issues associated with the selection of prior distributions. For a given problem, data cloning requires a full Bayesian model with proper prior distributions for all model parameters. The difference between data cloning and traditional Bayesian methods is that data cloning employs the likelihood function for K copies of the observed data
Under suitable regularity conditions, Walker (1969) showed that as the sample size increases, the posterior distribution converges to a multivariate normal (MVN) distribution with the MLE as the mean vector and the inverse of the Fisher information matrix as the covariance matrix. It means that the posterior distribution converges to the asymptotic distribution of the MLE, and for large sample sizes, the likelihood and Bayesian inferences become similar to each other.
If we assume that the K copies are independent of each other, the likelihood function of the K copies is given by 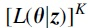 , where is exactly same as the maximum point of , where
, where is exactly same as the maximum point of , where
Let and the prior distribution
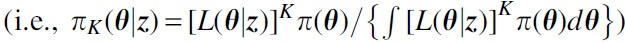 . Lele et al (
. Lele et al (The proof of Lele et al (2010) does not directly derived from the results of Walker (1969) because we cannot have independent K copied data sets in reality. It considers
If we use this result called data cloning, we do not have to solve complex optimizations to obtain the MLEs and their asymptotic SEs. We can generate samples of the K-cloned posterior,
3. METHODS
3.1. Bayesian model
In general, ML estimations for the ODE model in Equation (2) require to solve multiple optimization problems iteratively because it needs to estimate both ODE parameters and nuisance parameters. Since ODEs are inflexible models and gene expression data are noisy, it is not easy to solve the optimizations and it might fail to find the global optimum. To overcome this problem, we propose an estimation method for the ODE model of Equation (2) through data cloning.
As mentioned in Section 2.2, data cloning requires a full Bayesian model. To construct a full Bayesian model for Equation (2), we first derive the likelihood function for time-course gene expression data. Since the VAR model with lag one and MVN distribution are assumed in the ODE model of Equation (2), the likelihood function is given by
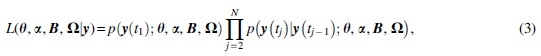
where
Now, we need to specify the prior distributions for the ODE parameter vector
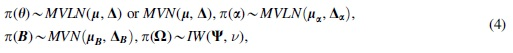
where MVNL represents a multivariate log-normal distribution and IW indicates an inverse Wishart distribution. As a prior distribution for the ODE parameter vector
From the likelihood function and prior distributions, we can have the posterior distribution for the original data set as follows:
where
3.2. Estimation through data cloning
Suppose that we independently repeat the same gene experiment K times and we obtain independent K data sets with the same values as the original time-course data  . Notice that, of course, we cannot have such independently repeated gene experiments in reality and the data cloning does not assume such independent experiments as mentioned in Section 2.2.
. Notice that, of course, we cannot have such independently repeated gene experiments in reality and the data cloning does not assume such independent experiments as mentioned in Section 2.2.
The posterior distribution for the K-cloned data is given by
Let
Equation (7) means that as
The posterior distribution
It is not easy to increase the sample size due to the cost and time for experiments, but we can easily increase the number of clones K. As shown in Equation (7), when K is large enough, the posterior distribution is almost degenerated and we can have the MLEs and their SEs closer and closer to the true estimates. Hence, to find an adequate number K, Lele et al (2010) proposed to use K when the ratio of the largest eigenvalues of the posterior covariance matrix from K-cloned data to that from the original data is less than a threshold. We follow their rule in the simulation and real data analysis.
4. SIMULATION
To verify the performance of the proposed estimation method, simulation* is performed in this section. In the simulation, we generate time-course data from the model of Equation (2). For the simulation, the following ODEs for three genes are considered:
Equation (8) presents a nonlinear ODEs for three genes. The specific setting for the model of Equation (2) is as follows: The gene expression levels at the initial time  , ODE parameters
, ODE parameters

True ODE trajectories and fitted ODE trajectories with data cloning. ODE, ordinary differential equation.
To verify the invariance property for the selection of prior distributions, we consider both informative and noninformative prior distributions. For the informative priors, Equation (4) is considered. To evaluate the accuracy of SE estimates for MLEs, we obtain an SE estimate for MLE of each ODE parameter using the Monte Carlo (MC) method. In the MC method, we estimate the MLE for each simulated data set by directly solving the maximization problem for the likelihood function of Equation (3), and then we obtain the SE estimate by computing standard deviation of 200 MLEs. In addition, we compute the coverage probabilities (CPs) for 90% and 95% confidence intervals (CIs) to investigate the accuracy of statistical inference from the proposed method.
For each simulated time-course data set, we obtain the MLE and its asymptotic SE estimate using the proposed data cloning method. Based on the MLE and its asymptotic SE estimate, we can construct the CI under the asymptotic normality. By counting the number of times that the CI includes the true parameter value among 200 CIs, we can compute CP. In this simulation, the performance of the proposed method is evaluated in terms of bias, SE, and CP.
Table 1 shows the mean of ODE parameter estimates, their biases in absolute value, the mean of their SEs, and 90% and 95% CPs obtained from the proposed data cloning method with informative and noninformative prior distributions, respectively. As shown in Table 1, for all statistics, there are no significant differences between informative and noninformative priors. It demonstrates that the proposed method meets the invariant property for the selection of prior distributions.
Simulation Results for the Proposed Data Cloning Method
CP, coverage probability; DC, data cloning; Infor., informative; MC, Monte Carlo; NInfor., noninformative; SE, standard error.
For both N = 30 and 50 cases, the biases of the ODE parameter estimates are relatively small and the estimated asymptotic SEs are also close to the SEs from the MC method. For both the ODE parameter estimates and asymptotic SEs, the differences slightly decrease as N increases. For accurate statistical inference, the CP should be greater than or equal to the confidence level. As shown in Table 1, although there exist many CP values less than their confidence level for N = 30 case, all CP values for N = 50 case are greater than or equal to their confidence level.
5. REAL DATA ANALYSIS
In this section, we estimate and infer parameters of an ODE model for a retina cell gene network of zebrafish using the method proposed in this study. The zebrafish eye anatomy is similar to that of humans, and its retina cells can restore vision. Thus, the GRN of zebrafish retina cells has been studied to develop treatments for impairment of human eye (Bibliowicz et al, 2011; Fumitaka et al, 2007; Qin et al, 2009).
Linder and Rempala (2013) proposed a network for three important genes,
In Equation (9), ODE parameters
Table 2 provides the MLE, asymptotic SE, and 95% CI for each ODE parameter, and Figure 2 shows data points and the fitted lines obtained using informative and noninformative prior distributions. As shown in Table 2 and Figure 2, the outcomes of informative and noninformative prior distributions are strikingly similar. Similarly to the synthetic data analysis in Section 4, it confirms that the inference by the proposed method is invariant for the choice of prior distributions.
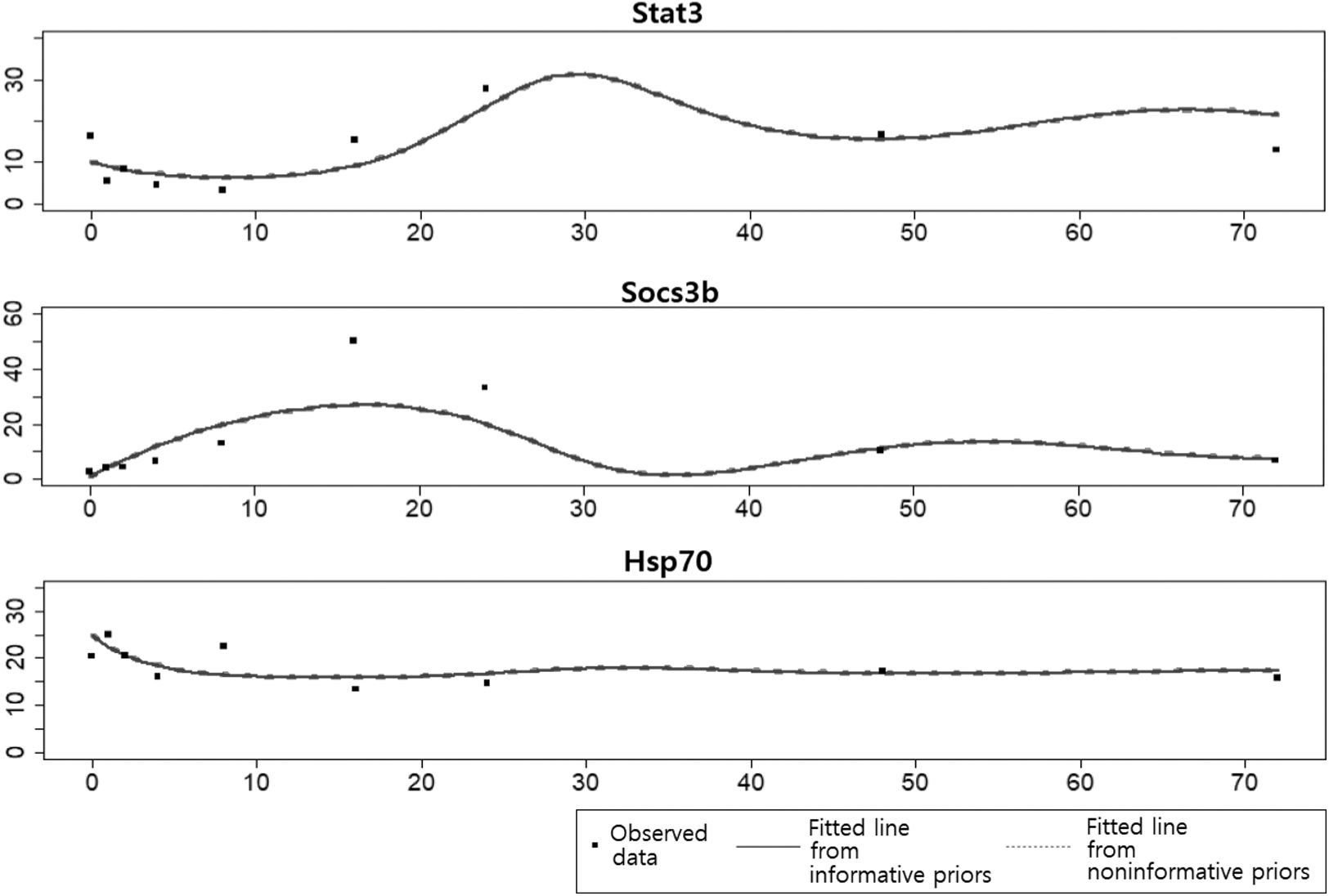
Data points and fitted ODE trajectories.
Estimation of Ordinary Differential Equation Parameters for Zebrafish Retina Cell Data
95% CI, 95% confidence interval; SE, standard error.
To infer the hypothesized GRN, we need to see the MLEs and 95% CIs for
6. SUMMARY AND DISCUSSION
ODE models are often used to describe the dynamics of GRNs and it is critical to understand whole gene regulation processes. For accurate estimation of ODE models for GRNs, we should consider intrinsic attributes of time-course gene expression data such as heteroscedasticity, correlation between genes, and time dependency. However, it is not easy to accurately estimate ODE models with such complex error structures. ML estimation approaches for such models could have local optimum problems due to iterative optimizations, and Bayesian estimation could have sensitivity issues depending on the choice of prior distributions due to small size of data. To overcome these problems, this study proposes an estimation method based on data cloning, which is an ML method through Bayesian MCMC. Since it uses Bayesian framework, it is free from local optimum problems, and its inference is invariant for the choice of prior distributions.
However, similar to Bayesian methods for complex models, the proposed method still requires high computing cost for the convergence of MCMC. To reduce the computing cost, we can consider sequential Monte Carlo (SMC) algorithms, also known as particle filters (Del Moral et al, 2006; Doucet et al, 2000). SMC methods combine importance sampling and resampling algorithms. To obtain posterior distributions, it sequentially samples from a sequence of intermediate probability distributions defined on a common space. It is expected to efficiently obtain target distributions.
Footnotes
AUTHORs' CONTRIBUTIONS
Methodology, software, formal analysis, investigation, data curation, and writing—original draft by D.S. Conceptualization, methodology, validation, writing—original draft, writing—review and editing, supervision, project administration, and funding acquisition by J.K.
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (Nos. NRF-2018R1D1A1B07049818 and NRF-2022R1F1A1072444).