
Abstract
The colored de Bruijn graph is a variation of the de Bruijn graph that has recently been utilized for indexing sequencing reads. Although state-of-the-art methods have achieved small index sizes, they produce many read-incoherent paths that tend to cover the same regions in the source genome sequence. To solve this problem, we propose an accurate coloring method that can reduce the generation of read-incoherent paths by utilizing different colors for a single read depending on the position in the read, which reduces ambiguous coloring in cases where a node has two successors, and both of the successors have the same color. To avoid having to memorize the order of the colors, we utilize a hash function to generate and reproduce the series of colors from the initial color and then apply a Bloom filter for storing the colors to reduce the index size. Experimental results using simulated data and real data demonstrate that our method reduces the occurrence of read-incoherent paths from 149,556 to only 2 and 5596 to 0 respectively. Moreover, the depths of coverage for the reconstructed reads are equal to those for the input reads for the simulated data, whereas the previous method decreases the depth of coverage at many positions in the source genome. Our method achieves quite a high accuracy with a comparable construction time, peak memory size, and index size to the previous method.
INTRODUCTION
The de Bruijn graph (dBG) is a data structure that is widely utilized for genome assembly (Pevzner et al., 2001). The colored de Bruijn Graph (cdBG) is a variation of dBG in which each node and edge can store multiple colors. The traditional dBG assumes that all input reads are generated from the same genome, whereas the cdBG can differentiate source genomes and generate contigs associated with different genomes by traversing the nodes storing the same color.
The cdBG was originally introduced for reference-free detection of a genetic variation among a small number of individuals (Iqbal et al., 2012) and has more recently been applied for indexing all the input reads to improve the accuracy of the variation detection (Alipanahi et al., 2018a,b; Díaz-Domínguez, 2019). A similar attempt for disambiguating a large number of paths in the graph has also been examined for a general variation graph constructed from VCF (Pandey et al., 2021).
A modern sequencer produces a huge number of sequencing reads, so reducing the memory consumption for dBG is crucial. Currently, BOSS (Bowe et al., 2012) is widely accepted as a memory-efficient form of dBG, and it is also used as a part of cdBGs (Almodaresi et al., 2019; Almodaresi et al., 2017; Alipanahi et al., 2018a,b; Díaz-Domínguez, 2019; Holley et al., 2015; Muggli et al., 2017).
For example, VARI (Muggli et al., 2017) uses BOSS for a graph and stores the colors of nodes and edges in a binary matrix C, where rows represent k-mers and columns represent colors by using Elias-Fano encoding (Elias, 1974; Fano, 1971; Okanohara and Sadakane, 2007). C is held as a compressed form to reduce memory use. As most other memory-efficient cdBGs take a similar approach to VARI, designing an efficient C is essential, particularly when a cdBG supports read-coherent paths (i.e., when it assigns colors to differentiate each read).
To reduce the size of C, previous studies (Alipanahi et al., 2018b; Díaz-Domínguez, 2019) have reused colors in a greedy manner, and although this is successful in terms of reducing the index size, there is still a problem in that subgraphs causing read- incoherent paths are generated. More precisely, it is not possible to uniquely determine the next node when traversing the subgraphs, which leads to a failure to reproduce some input reads that overlap the subgraphs. We call such an input read an ambiguous sequence.
An example is shown in Figure 1, where the subgraphs correspond to three input reads r1 (embedded as a path:
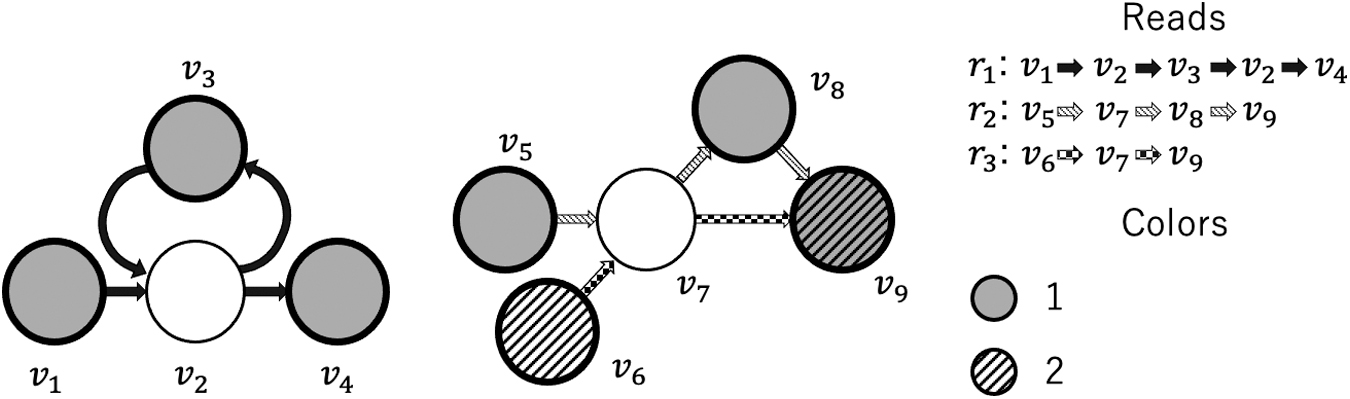
Coloring examples of previous method. Reads r1 and r2 cannot be reconstructed because v2 and v7 have two successors recording the same color.
We traverse the graph in such a way that we choose nodes that have the same color to find a read-coherent path. Therefore, if we want to find a path that is coherent with a read, we should traverse the nodes that are storing the color of the read. That is, if we encounter a node with more than one successor, the node storing the color of the read should be chosen.
The problem with the coloring scheme of prior works is that it tends to generate subgraphs in which more than one successor have the same color, as illustrated by the subgraphs in Figure 1. For example, the same color is recorded at v3 and v4, which are the successors of v2. v8 and v9 are the successors of v7 and they have the same color. The ambiguity is caused by the underlying structure of the source genome sequence. In the subgraph on the left, r1 contains a repeat
Generally, reads originating from the same region in the source genome overlap the same subgraph, so if a region causes such ambiguity, all the reads covering the region potentially become ambiguous sequences when they are embedded in the cdBG. This means that no read-coherent path can be found by cdBG for such regions. To demonstrate the problem, we show in Figure 2 a case in which all the reads covering a particular region are ambiguous sequences. In fact, we found many such information-lost regions (discussed further in Section 3). Since the regions causing the ambiguity may contain biologically meaningful structural variants, designing a coloring scheme that can avoid producing ambiguous sequences will contribute to the accuracy of the downstream analysis.

Example of an information-lost region in the source genome. The horizontal axis shows the position in the source genome and the vertical axis shows the number of reads that support that position. The number of ambiguous sequences increases as it gets closer to the repeat region at the center of the figure, and there are no non-ambiguous sequences in the repeat region.
In this article, we introduce a novel coloring algorithm for cdBG that can index all the input reads accurately. In this algorithm, multiple colors are used for a single read to avoid the ambiguity. Although our approach increases the number of colors, it keeps the index size small by using a hash function-based color generation technique and a Bloom filter. Our experimental results demonstrate that the proposed algorithm reduces the occurrence of ambiguous sequences from 149,556 to only 2 for simulated data and 5596 to 0 for real data while achieving a similar build time, peak memory size, and index size to the previous method.
In this section, we explain our coloring algorithm in detail. First, we provide definitions and description of related algorithms in Section 2.1. We then explain our concept of coloring based on hash functions in Section 2.2, followed by the detailed construction procedure and the search algorithms in Sections 2.3 and 2.4. Finally, we describe how to compress a color matrix in Section 2.5.
Preliminaries
Notations
Given a string S, we denote the i-th letter of S by
Rank and select
Given a string Q of length n that consists of the alphabet
de Bruijn graph
A dBG is a labeled directed graph 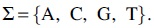 . In this study, R consists of a set of reads
. In this study, R consists of a set of reads
In principle, our algorithm can use any form of dBG as long as any node in dBG is uniquely identified. We use BOSS (Bowe et al., 2012) in this study to take advantage of its memory efficiency. BOSS (Bowe et al., 2012) is a succinct representation of dBG and is based on an idea similar to BWT (Burrows and Wheeler, 1994). It performs various graph operations within an efficient computation time (e.g., the numbers of outgoing and incoming edges of a node can be computed in constant time) while consuming only a small amount of memory. BOSS supports the following operations.
In addition to the above, our algorithm implements the following operations that are used in the previous research (Díaz-Domínguez, 2019). These operations can also be computed efficiently.
Colored de Bruijn graph
A cdBG is an extension of the dBG. It records a set of colors at each node (or edge). The color c is added to a node v if the input read r is assigned the color c and the label of v is a
To reduce the number of columns in C, we categorize a node into four types—start, end, critical, and non-colored—and give colors only to the start, end, and critical nodes because the color of a non-colored node can be derived from its predecessor node, which is the same technique used in the previous study (Díaz-Domínguez, 2019). The start node is the node that represents the beginning of a read r.
The label of the node is $
Our algorithm needs the following operations on dBG and C. We will show how to implement them in Sections 2.3 and 2.5.
Concept of proposed coloring scheme
As described in Section 1, the previous approach generates ambiguous subgraphs in which a node has several successors storing the same color. A simple way to solve this problem is to use different colors for a single read depending on the position in the read, so that we can choose a correct node. Consider the case where a read is CATATC and k = 3, and the topology is that of the left graph in Figure 1. v1,
If we assign the i-th 2-mer a color i (e.g., CA is assigned 0 and AT is assigned 1), the colors of v1, v2, v3, and v4 become 0, {1, 3}, 2, and 4. Starting from v1, we can traverse
To maintain the order of the colors without using an additional index, we utilize a hash function to generate a series of colors such that the color of the node to be visited next can be reproduced from the color of the current node.
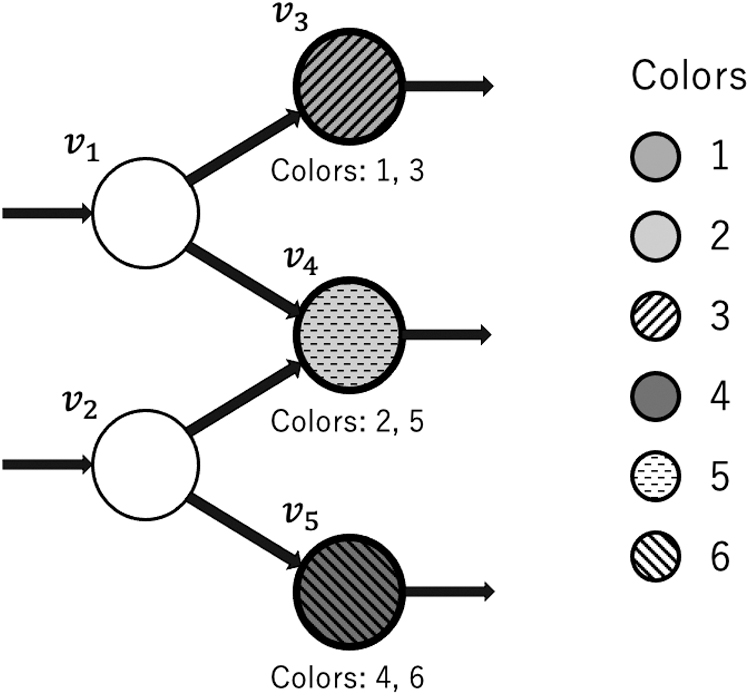
To introduce the key idea, we use a toy example shown in Figure 3. In our algorithm, we assume the topology of the graph and the type of each node are given before assigning colors. For the toy example, we assign color 1 to read r1. The series of colors used for embedding r1 is determined by a hash function in such a way that a next color is computed by a current node ID and a current color. In the first step, we record 1 at v1 because r1 starts at v1 and the color of r1 is 1. In the second step, we compute a new color

Example of coloring by our method for the same graph as Figure 1. Start nodes v1, v5, and v6, end nodes v4 and v9, and critical nodes v3 and v8 need to be colored. To embed r1 on the graph, we visit
As mentioned in the previous section, we do not color all the nodes but only the start, critical, and end nodes. Therefore, we do not color v2 and we store a color
Our method begins by constructing a BOSS G of an input read set R and scanning G to determine the type of each node. It then traverses on G for all the reads and counts how many times each node is visited if the node is either a start, end, or critical node. We denote the number of colored nodes by p and a count for the i-th colored node by
As described in Section 2.1, C is an
To identify the set of colors of an i-th colored node, we use D, which is a concatenation of
In our coloring method, a series of colors for embedding a read is added read by read. In Line 3 of Algorithm 1, a terminal symbol $ is added at the head and tail of a read r so that we can distinguish the beginning and end of the read. In Line 4, the start node v for r is obtained and we compute the order of v within the colored nodes in Line 5. In Line 6, a set of colors T already assigned to v is obtained, and we compute
In Line 7, the position of the first 0 (i.e., the first position of empty elements) within the range is obtained by a binary search and c is stored at the position in Line 8. In Line 9, the next color to be used is computed by hash 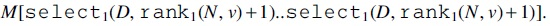 .
.

Search on the color matrix
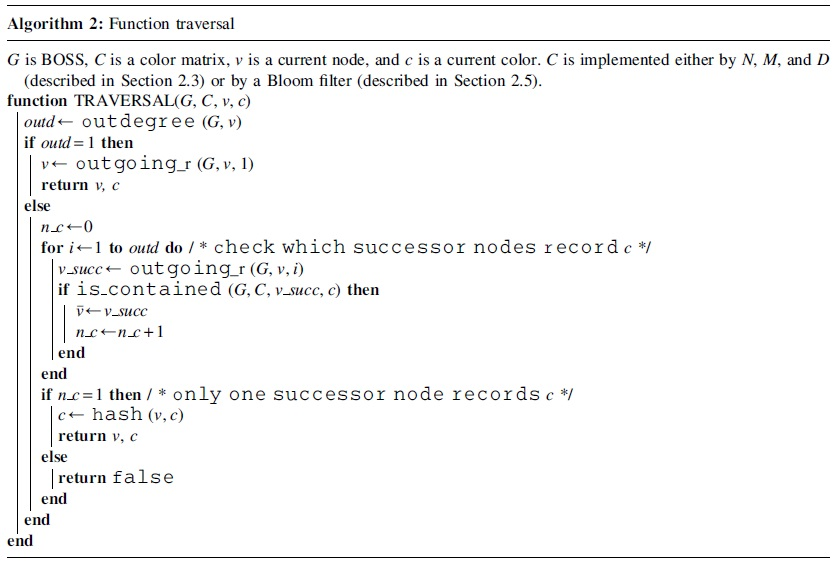
Once we construct a color matrix C, we can easily find a read-coherent path. Algorithm 2 describes a traverse. Given G, C, a node v, and a color c, it returns the next node and color. Note that it returns false when the next color is contained in more than one successor of v, as we cannot uniquely determine the next node. Such a case could happen due to a hash collision. For example, as shown in Figure 4, if two reads r1 and r2 share a path

Examples of ambiguous sequences in our method. The upper graph shows an example where the same hash value is generated at the same node v1 for reads r1 and r2, and the same color is recorded at each successor node, resulting in ambiguity. The lower graph shows an example where the same hash value is generated at v5 and v8 and stored at v7 and
The other example with r3, r4, and r5 is caused by a similar reason. Although such a collision generates ambiguous sequences, we should emphasize that a hash collision happens randomly, so the probability of such an ambiguity occurring at the same node is quite small. This means that, since an ambiguous read covers a random position in the source genome, we can maintain the depth of read coverage for any position in the source genome—unlike the previous method, which tends to lose all the read-coherent paths for a particular region. The occurrence of a hash collision can be mitigated if we use a hash function that maps to a large space; however, this causes an increase in the size of M.
In the previous section, we explained the color matrix consisting of N, D, and M. Since our algorithm needs several colors to embed each read, M becomes large, particularly when the colors are randomly distributed over a large space. In our implementation, we used 64 bits for each color to avoid a hash collision. After constructing the color matrix, M is utilized to check whether a given color is contained in a given node, and we do not have to hold all the colors explicitly. In this subsection, we propose a space efficient method to enable the membership query while achieving a small false positive rate.
The key detail of the membership query in our case is that we know all the potential queries for a given node v, since we already know the topology of the graph and the set of colors stored in each node. That is, when we visit a node with multiple outgoing edges, the color that is used for selecting the next node is always stored in one of the successors, as per the definition of our coloring scheme. Let us define a set
Example of
We use a Bloom filter (Bloom, 1970) for the membership query (see Section 1 of the Supplementary Material S1 for a brief explanation of the Bloom filter). To reduce the filter size, we examine the length of a bit vector and the number of hash functions from a small value toward a large value until the ones that achieve a false positive rate
Scan cdBG and obtain
Determine the number of hash functions as
Add all the colors in
Check membership of all the colors in
Build the filter if the false positive rate is less than or equal to p; otherwise, set
Note that this simple method can achieve a smaller filter size compared to the approach based on minimal perfect hash function. We find
See Section 2 of the Supplementary Material S1 for a more detailed discussion. Note that the bit lengths are slightly different from the values presented in Section 3, since we need to assume the case where nodes with
In the compressed form of C, the function is_contained
In our method, the i-th hash function is implemented by
Figure 6 shows the summary of the color matrix. As shown in the figure, the position in B for the filter of i-th color node is extracted by
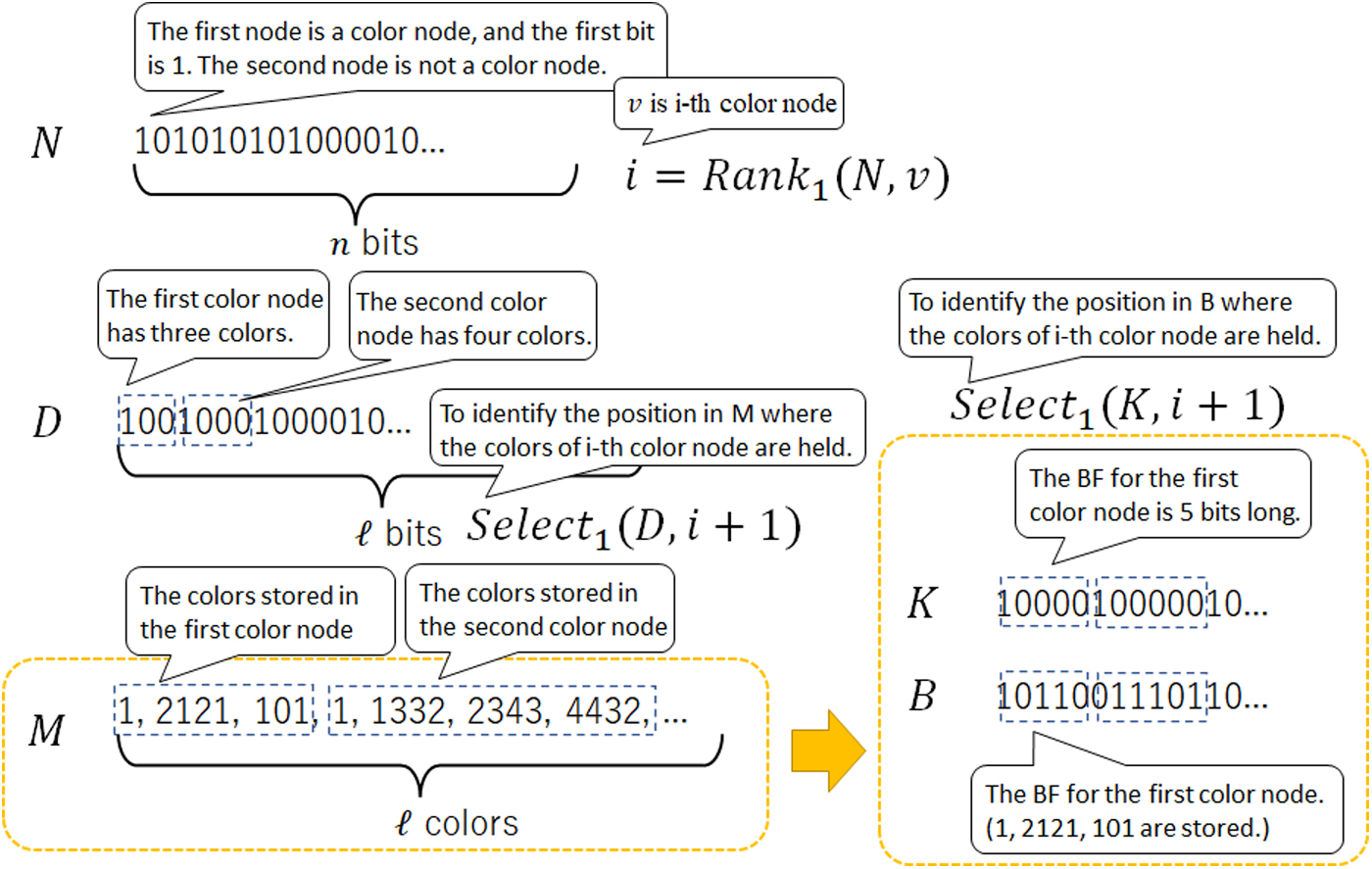
The summary of the color matrix. After obtaining the node id v from dBG, the colors stored in v are computed using N, D, and M. M, which directly holds colors, is replaced by the Bloom filters B, which is more space-efficient. The filter for each color node is extracted by K.
Given a start node v and an initial color c0, we can find a read-coherent path by running Algorithm 2. Although c0 is not explicitly given, it can be found by using v's filter. In our construction algorithm, the initial color assigned to each read is the minimum number except for the colors already stored. The colors assigned to critical and end nodes are generated by a hash function. Since the hash function maps to a large space, the initial color (i.e., the color assigned to a start node) is usually 1.
Therefore, we know the initial color by checking the membership of 1 in v's filter. If v is a start node of multiple reads, the initial colors for the reads become
We implemented our algorithm and compared it with a state-of-the-art method (Díaz-Domínguez, 2019) to evaluate the performance. Since a suffix tree (Sadakane, 2007) is often used for indexing genome sequences and we can perform the necessary operations for finding a read-coherent path, we also compared our algorithm with the suffix tree. Note that using only FM-Index for finding a read-coherent path is quite time consuming, so we did not make a comparison with FM-Index.
Data sets and implementation
We tested our method both on simulated data and real data. The simulated read sets were generated from chromosome 21 of the human reference genome (GRCh38) using Art (Huang et al., 2012). We utilized the HiSeq2000 error profile and generated 100 bp single-end reads. The number of reads was
Since our algorithm indexes the reverse complements of the input reads,
For the real read sets, we downloaded the whole genome sequencing data of Escherichia coli (ERR4785033) from NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) and excluded reads including N. We call this data Realdat 1. The number of reads for Realdat 1 was
We implemented our algorithm and the suffix tree by using SDSL-lite library (Gog, 2013). The hash function is implemented similarly to the one in Boost (Boost.org, 1999) that is based on the computationally efficient algorithm (Wheeler and Needham, 1994) (see Listing 1 in the Supplementary Material S1). All the programs were compiled with the -O3 optimization option and run on a machine equipped with Intel(R) Core(TM) i7-7820X CPU@3.60GHz and 125GB of RAM.
For each method, we built indices and measured the execution time, peak memory, and index size. Since our method and the previous method support multiple threads for building an index, we used 16 threads. The dBG was constructed with 30-mer for both methods, and the suffix tree was built by a single thread. For our method, we tested three different parameter settings: #1
Results on simulated data
The results of the index construction are shown in Table 1, where we can see that our method achieved an index building time, peak memory, and index size comparable to those of the previous method. There was a trade-off between the index building time and the index size depending on the setting of ds in our method.
Results of Index Construction for the Simulated Read Sets
Results of Index Construction for the Simulated Read Sets
We examined three different parameter settings: #1, #2, and #3. The building time of entire index includes that of C, and the entire index size also includes the size of C
When we used settings #2 or #3, our method achieved the smallest index size. The suffix array generated an index more than five times larger than our method and the previous method, though its index building time was small. When we counted the number of colors (i.e.,
To evaluate the efficiency of the constructed indices, we reconstructed all the input reads and their reverse complements (i.e.,
Results of Reconstructing Original Reads from the Indices for the Simulated Read Sets
The peak memories are generally proportional to the index size, but the reconstruction time differs significantly between the previous method and ours. Since we set the false positive probability
The peak memories of our method and the previous method are comparable, and that of the suffix tree was twice as large as our method and the previous method. The reconstruction time of our method was significantly faster compared to the previous method. In the previous method, the membership query is conducted in such a way that all the colors in a node v are reconstructed and then scanned, which requires a time linear to the total number of colors stored in nodes to be visited.
In contrast, our method conducts the membership query more efficiently by using a Bloom filter, which only requires a time linear to the number of hash functions. We note that other state-of-the-art cdBGs (Almodaresi et al., 2017; Muggli et al., 2017) also require the constant number of rank/select operations on the compressed data structures for finding a color in a node. The time difference between our method and the Suffix tree can be interpreted as the overhead of finding colors.
We examined three different settings for Bloom filters, that is, three different
We also computed the depth of the coverage for the reconstructed reads at all the positions in chromosome 21 of the human reference genome except for N (
Results of Number of Bases in Chromosome 21 of the Human Reference Genome Except for N (
With our method, the results were the same as the original read sets used as input to create indices, but with the previous method, the depth became shallow for many bases.
We also conducted the experiments using real read sets. Table 4 shows the results of the index construction. Overall, the results show a similar tendency that was observed in the results of the simulated data, except that the results of our methods based on #2 setting (
Results of Index Construction for the Real Read Sets
Results of Index Construction for the Real Read Sets
We examined three different parameter settings: #1, #2, and #3. The building time of entire index includes that of C, and the entire index size also includes the size of C.
For the case using #3 setting, the filter construction cost becomes high for a node storing a large number of colors that has siblings storing many colors, which could be a computational bottleneck, especially when the number of colors stored in siblings is large (i.e.,
Our method achieved the smallest index size, while the peak memory size of the previous method was the smallest. Realdat 2 is an error-corrected read set, which includes a slightly smaller number of reads and is expected to include a smaller number of errors that may cause an increase in the number of nodes and colors. The results also show that the building time, the peak memory size, and the index size of Realdat 2 are smaller than those of Realdat 1 for all the methods.
We also reconstructed all the input reads and their reverse complements and summarized the results in Table 5. Our methods generated no ambiguous sequences, whereas the previous method generated several thousand ones. Note that we cannot evaluate the depth of coverage for each position in the reference genome because we cannot know the true positions where each read was sequenced.
Results of Reconstructing Original Reads from the Indices for the Real Read Sets
In this work, we developed a new method for indexing sequencing reads by a cdBG. The proposed method uses different colors for a single read depending on the position in the read to reduce the generation of ambiguous subgraphs such as two successor nodes having the same color. To avoid having to memorize the order of the colors, we utilized a hash function to generate colors so that the series of colors can be reproduced from the initial color.
We also proposed using a Bloom filter for holding a set of colors at a node to reduce the index size. We implemented our method and compared it with the previous method and a suffix tree by using simulated data and real data. The results showed that our method significantly reduced the generation of ambiguous subgraphs, and the number of input reads that could not be reconstructed was at most only 10 for simulated data and 0 for real data, whereas the previous method could not reconstruct at least 149,556 reads for simulated data and 5596 reads for real data.
We also confirmed that the depths of coverage for the reconstructed reads were equal to those for the input reads when using our method, whereas the previous method decreased the depth of coverage at many positions in the source genome. Our method achieved such a high accuracy with a comparable construction time, peak memory size, and index size to the previous method, and the index size was one-fifth that of the suffix array. In addition, the membership check of a color in our method requires only a time linear to the number of hash functions used in a Bloom filter and does not depend on the bit length of colors stored in a node.
One of the future works is to extend the method to a dynamic cdBG, considering the growing interest in an updatable data structure (Holley and Melsted, 2020; Muggli et al., 2019). The potential approach is to use a dynamic dBG (Alipanahi et al., 2021) instead of BOSS, which still enables a correct graph traversal after deleting a node; however, after adding a node, it is necessary to change the colors of nodes around the node, which may require a high computational cost. Although some challenges remain, our findings demonstrate the efficiency of our method, and we believe it will contribute to the analysis of large-scale sequencing data.
Footnotes
ACKNOWLEDGMENT
The authors thank Mr. Taiki Yamada for fruitful discussions.
AUTHORs' CONTRIBUTIONS
N.H.: conceptualization, methodology, software, writing—original draft. K.S.: conceptualization, methodology, writing—review and editing, supervision.
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
The part of the research is supported by Kayamori Foundation of Informational Science Advancement.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.