
Abstract
Metagenomics enables the recovery of various genetic materials from different species, thus providing valuable insights into microbial communities. Metagenomic binning group sequences belong to different organisms, which is an important step in the early stages of metagenomic analysis pipelines. The classic pipeline followed in metagenomic binning is to assemble short reads into longer contigs and then bin these resulting contigs into groups representing different taxonomic groups in the metagenomic sample. Most of the currently available binning tools are designed to bin metagenomic contigs, but they do not make use of the assembly graphs that produce such assemblies. In this study, we propose MetaCoAG, a metagenomic binning tool that uses assembly graphs with the composition and coverage information of contigs. MetaCoAG estimates the number of initial bins using single-copy marker genes, assigns contigs into bins iteratively, and adjusts the number of bins dynamically throughout the binning process. We show that MetaCoAG significantly outperforms state-of-the-art binning tools by producing similar or more high-quality bins than the second-best binning tool on both simulated and real datasets. To the best of our knowledge, MetaCoAG is the first stand-alone contig-binning tool that directly makes use of the assembly graph information along with other features of the contigs.
1. INTRODUCTION
With the emergence of high-throughput sequencing technologies, metagenomics has facilitated the analysis of microbial communities without the need for culturing, especially in large scale metagenomics studies such as the Human Microbiome Project (Turnbaugh et al., 2007) and the Metagenomics and Metadesign of the Subways and Urban Biomes (Mason et al., 2016). These microbial communities consist of a large number of micro-organisms, including various beneficiary and pathogenic bacteria. Large amounts of sequencing reads that originate from the underlying micro-organisms of an environmental sample can be obtained by sequencing the sample directly. This enables the recovery of genetic material from many micro-organisms, especially those that cannot be grown in standard laboratory media.
Characterizing the composition of an environmental sample and identifying the micro-organisms that are present enable downstream analysis of the behavior and functions of microbial communities. To facilitate this analysis, we perform metagenomic binning, where we cluster sequences into bins that represent different taxonomic groups such as species, genera, or higher levels (Sedlar et al., 2017).
Next-generation sequencing technologies such as Illumina allow us to sequence microbial communities and obtain highly accurate short sequences called reads. Binning these reads before assembly (Cleary et al., 2015; Ounit et al., 2015; Vinh et al., 2015; Girotto et al., 2016; Alanko et al., 2017; Schaeffer et al., 2017; Luo et al., 2018) can produce less reliable results due to the short read lengths (Yu et al., 2018). Hence, the popular pipeline followed during metagenomic analysis is to first assemble short reads into longer sequences called contigs and then bin these assembled contigs into groups that represent different taxonomic groups (Sedlar et al., 2017). These bins of contigs enable the construction of metagenome-assembled genomes (MAGs) that represent complete partial microbial genomes (Yang et al., 2021).
The latest contig-binning approaches fall into two broad categories (Yue et al., 2020): (1) reference-based binning approaches (Wood and Salzberg, 2014; Ounit et al., 2015; Kim et al., 2016; Menzel et al., 2016), which classify contigs with labels of known taxonomic groups by comparing against a reference database, and (2) reference-free binning approaches, which cluster contigs into unlabeled bins based on genomic features of these contigs. Early microbiome studies have relied heavily on reference-based approaches for taxonomic assignments (Yang et al., 2021). However, currently available reference genomes may be incomplete or have low quality, and reference genomes of previously uncharacterized micro-organisms may not be available in current databases. Hence, reference-free binning approaches (Strous et al., 2012; Alneberg et al., 2014; Wu et al., 2014, 2015; Lin and Liao, 2016; Lu et al., 2016; Laczny et al., 2017; Yu et al., 2018; Kang et al., 2019; Wang et al., 2019; Wickramarachchi et al., 2020; Nissen et al., 2021; Chandrasiri et al., 2022) have become popular as they enable the identification of new species without the need to compare against reference databases.
Reference-free contig-binning tools mainly make use of two main features to perform binning: (1) composition, obtained as normalized frequencies of oligonucleotides of length k (referred to as k-mers) and (2) coverage, considered as the average number of reads that map to each base of the contig. These tools achieve improved performance by combining both the composition and the coverage information, and different tools follow different algorithmic approaches to identify bins and place contigs in these bins. Recently published tools such as Vamb (Nissen et al., 2021), LRBinner (Wickramarachchi and Lin, 2021, 2022b), and RepBin (Xue et al., 2022) have successfully applied machine learning techniques to capture the species-specific signals of sequences into a low-dimensional space that facilitates efficient clustering. However, it still remains challenging for these binning tools to accurately reconstruct microbial genomes of species with similar composition and coverage profiles.
Estimating the number of species present in a given sample is another major challenge in metagenomic binning. Recent binning tools have made use of single-copy marker genes [special marker genes that appear only once in the genome and are conserved in the majority of bacterial genomes (Dupont et al., 2012; Albertsen et al., 2013; Wu et al., 2014)] to estimate the number of species. The single-copy marker gene information is underutilized in binning tools such as MaxBin (Wu et al., 2014), MaxBin2 (Wu et al., 2015), and SolidBin (Wang et al., 2019) as these tools use only one marker gene to estimate the number of initial bins, which may lead to an underestimation of the number of species. Hence, it is worth investigating how to make use of multiple single-copy marker genes together to obtain a better estimate for the number of bins and to explore more features of contigs that can improve the binning process.
Contigs are produced by joining reads into longer sequences through a process known as assembly, and many tools have been developed to perform assembly. Special assemblers known as metagenomic assemblers have been developed to assemble metagenomic datasets. Most existing metagenomic assemblers (Peng et al., 2012; Li et al., 2015; Nurk et al., 2017) use assembly graphs as the key data structure [e.g., simplified de Bruijn graph (Pevzner et al., 2001)] to assemble reads into contigs. Previous studies indicated that contigs connected to each other in the assembly graph are more likely to belong to the same taxonomic group (Barnum et al., 2018; Mallawaarachchi et al., 2020a). Although popular metagenomic assemblers such as metaSPAdes (Nurk et al., 2017) output contigs along with their connection information in the assembly graph, most existing binning tools ignore the valuable connection information between contigs.
More recently, bin-refinement tools such as GraphBin (Mallawaarachchi et al., 2020a), GraphBin2 (Mallawaarachchi et al., 2020b, 2021), METAMVGL (Zhang and Zhang, 2021), and GraphPlas (Wickramarachchi and Lin, 2022a) have been developed to refine existing binning/classification results using assembly graphs. These tools rely upon the bins produced by an existing binning tool and cannot dynamically adjust the number of bins. Moreover, recently introduced metabinners such as DAS tools (Sieber et al., 2018) and MetaWRAP (Uritskiy et al., 2018) integrate and optimize the results of multiple binning approaches. Even though these tools achieve improved binning performance, they still require initial binning results obtained from other existing binning tools and some tools cannot dynamically adjust the number of bins. Hence, it is worth exploring methods to develop a stand-alone contig-binning tool that makes use of the assembly graph information along with the composition and coverage information of contigs.
In this study, we introduce MetaCoAG, a reference-free stand-alone approach for binning metagenomic contigs. In addition to composition and abundance information, MetaCoAG makes use of the connectivity information from assembly graphs for binning. To the best of our knowledge, MetaCoAG is the first contig-binning tool to make direct use of the assembly graph information. We benchmark MetaCoAG against state-of-the-art contig-binning tools using simulated and real datasets. We also test MetaCoAG using the simulated metagenome data from the toy Human Microbiome project of the second Critical Assessment of Metagenomic Interpretation (CAMI) challenge (Meyer et al., 2022). The experimental results show that MetaCoAG significantly outperforms other contig-binning tools, for example, improving the completeness of bins while maintaining high purity levels and producing more high-quality bins.
2. METHODS
Figure 1 illustrates the overall workflow of MetaCoAG where each step of MetaCoAG is explained in detail in the following sections.
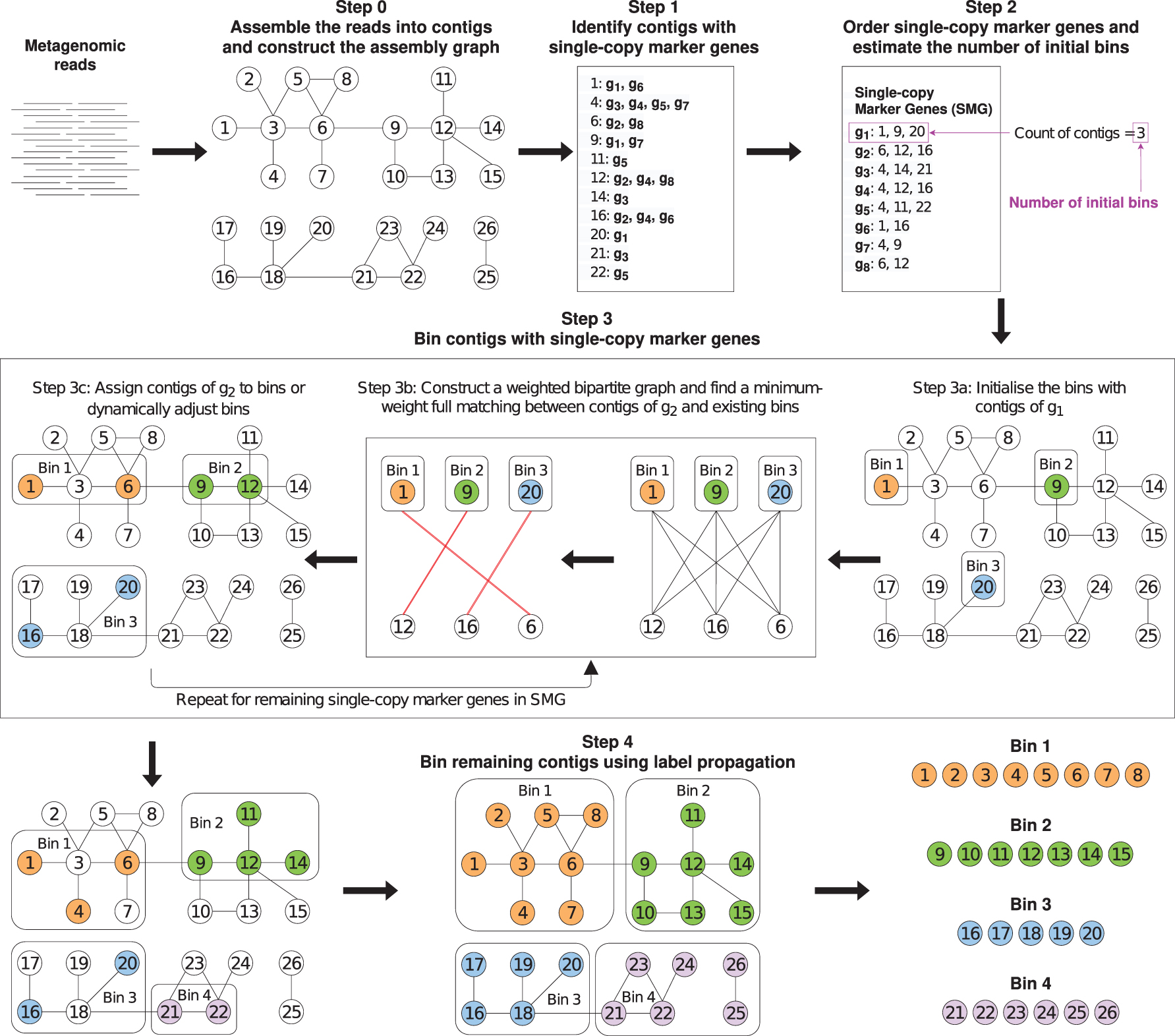
MetaCoAG workflow.
2.1. Step 0: Assemble reads into contigs and construct the assembly graph
A preprocessing step consists of assembling the reads into contigs using a metagenomic assembler and obtaining the assembly graph. Metagenomic assemblers first use graph models to connect overlapping reads or k-mers and infer contigs as nonbranching paths. After graph simplification, the vertices represent contigs and edges represent connections between contigs in the assembly graph. Here, we use the popular metagenomic assembler metaSPAdes (Nurk et al., 2017) to derive the contigs and assembly graph, which are used as inputs to MetaCoAG. Note that the assembly graphs can also be obtained similarly using other metagenomic assemblers such as MEGAHIT (Li et al., 2015) and metaFlye (Kolmogorov et al., 2020).
2.2. Step 1: Identify contigs with single-copy marker genes
Single-copy marker genes are special marker genes that appear exactly once in a bacterial genome and are conserved in the majority of bacterial genomes (Dupont et al., 2012; Albertsen et al., 2013; Wu et al., 2014). We use FragGeneScan (Rho et al., 2010) and HMMER (Eddy, 2011) to identify the contigs, which contain each single-copy marker gene (as shown in Step 1 of Fig. 1). A single-copy marker gene is considered to be contained in a contig if more than 50% of the gene length is aligned to this contig. In an ideal assembly, a single-copy marker gene from one species should only be contained in one contig from this species.
Similar to approaches such as MaxBin (Wu et al., 2014), MaxBin2 (Wu et al., 2015), and SolidBin (Wang et al., 2019), MetaCoAG uses single-copy marker genes to distinguish contigs belonging to different species, that is, if multiple contigs contain the same single-copy marker gene, they should belong to different species, respectively.
2.3. Step 2: Order single-copy marker genes and estimate the number of initial bins
For a given single-copy marker gene, the contigs containing this marker gene should come from different species (e.g., if two contigs contain the same marker gene, then the two contigs should belong to two different species). In the ideal case, if we have a near-perfect assembly, the number of contigs that contain the same single-copy marker gene should be equal to the number of species present in the sample. However, in reality, assemblies can be fragmented and erroneous, which make it challenging to recover all single-copy marker genes and, hence, can lower the counts of contigs containing each single-copy marker gene.
To get a better estimate of the number of species, we obtain the counts of contigs containing each single-copy marker gene. We also record the single-copy marker genes found in each contig. For a single-copy marker gene, the number of contigs that it can distinguish is the number of contigs containing this gene. Therefore, we order all the single-copy marker genes according to the descending order of the number of contigs containing them (as shown in Step 2 of Fig. 1). We refer to this list of ordered marker genes as SMG, where a single-copy marker gene gi has a set of contigs
2.4. Step 3: Bin contigs with single-copy marker genes
2.4.1. Step 3a: Initialize bins
We initialize the bins using the contigs of the first single-copy marker gene g1 in SMG; that is, we initialize a new bin B for each contig in
2.4.2. Calculating composition and coverage similarities
Previous studies on metagenomic binning have used genomic signatures as they follow species-specific patterns (Deschavanne et al., 1999; Wu et al., 2014). The most commonly used genomic signatures to characterize composition information are tetranucleotide frequencies (136 canonical 4-mers, also known as tetramers; Alneberg et al., 2014; Wu et al., 2014, 2015; Kang et al., 2019; Wang et al., 2019; Nissen et al., 2021). For each contig c, we normalize the tetranucleotide frequencies using its total number of tetranucleotides to obtain the normalized tetranucleotide frequency vector,
We use the same formula proposed by Wu et al. (Wu et al., 2014) to estimate how similar c and

We use the coverage information of the contigs as coverage carries important information about the abundance of species and has been used in previous metagenomic binning studies (Albertsen et al., 2013; Wu et al., 2014; Kang et al., 2019; Wang et al., 2019; Nissen et al., 2021). Shotgun sequencing has shown to follow the Lander–Waterman model (Lander and Waterman, 1988) and the Poisson distribution has been used to obtain the sequencing coverage of nucleotides and applied in metagenomic binning (Wu and Ye, 2011; Wu et al., 2014). Modifying the definition found in Wu et al. (2014), we estimate how similar c and
Here,
2.4.3. Step 3b: Construct a weighted bipartite graph and find a minimum-weight full matching
In the previous steps, we have used single-copy marker genes to identify pairs of contigs that belong to different species. Remind that contigs in different bins in BINS are expected to belong to different species and contigs in
In Equation (3),
Now, we find a minimum-weight full matching (minimum-cost assignment; Karp, 1980) for the above bipartite graph between
In the next step, we will see how we can assign the contigs to existing bins based on the minimum-weight full matching we have obtained.
2.4.4. Step 3c: Assign contigs to existing bins and dynamically adjust bins
Previous studies have observed that contigs connected to each other in the assembly graph are more likely to belong to the same taxonomic group (Barnum et al., 2018; Mallawaarachchi et al., 2020a). While
We define the thresholds intraspecies weight
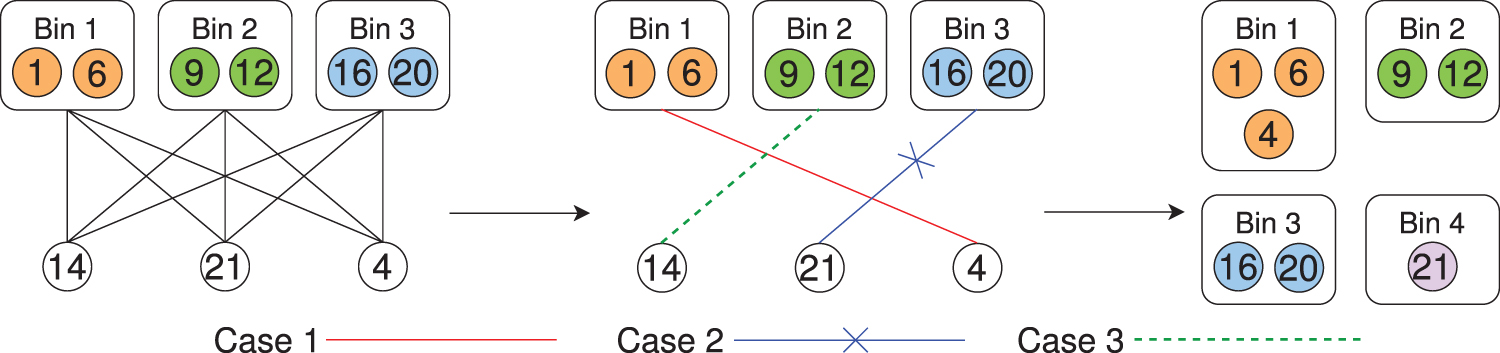
Cases 1, 2, and 3 in assigning contigs to existing bins or adjusting bins.
The default values for parameters
2.5. Step 4: Bin remaining contigs using label propagation
After we bin the contigs with single-copy marker genes, each such contig receives a label corresponding to its bin. Now, we will propagate labels from these contigs to other unlabeled contigs within the same connected component (as shown in Step 4 of Fig. 1).
2.5.1. Step 4a: Propagate labels within connected components
MetaCoAG uses composition, coverage, and distance information from the assembly graph to propagate labels from labeled contigs to the unlabeled contigs located within the same connected components. More specifically, for each unlabeled long contig c (at least 1000 bp long because short contigs result in unreliable composition and coverage information) directly connected or connected via short contigs to a labeled contig
MetaCoAG iteratively selects the candidate propagation action with the highest priority and executes the corresponding label propagation. If a contig to be labeled contains single-copy marker genes, the relevant candidate propagation action is executed if the single-copy marker genes of the contig are not present in the intended bin. We restrict the depth of the search for labeled contigs in this step to 10.
2.5.2. Step 4b: Propagate labels across different components
Note that some components in the assembly graph may not have any labeled contigs, and we need to propagate labels from labeled bins to unlabeled contigs across components. Calculating pair-wise weights
This propagation is limited to long contigs (at least 1000 bp long by default). If an unlabeled contig contains single-copy marker genes, it is assigned to bin B that minimizes
2.5.3. Step 4c: Postprocessing
In this step, we will make final adjustments on the current bins. Two bins B and
3. EXPERIMENTAL SETUP
3.1. Datasets and tools
3.1.1. Simulated datasets
We evaluated the binning performance on the simulated
3.1.2. CAMI2 toy human microbiome project datasets
We used the simulated metagenome data from the toy Human Microbiome Project dataset of the second CAMI challenge (Meyer et al., 2022). Metagenomes with HiSeq reads were simulated from five different body sites of the human host as follows.
Urogenital tract—referred as
Skin—referred as
Oral cavity—referred as
Gastrointestinal tract—referred as
Airways—referred as
3.1.3. Real datasets
We used the following three real datasets from the National Center for Biotechnology Information (NCBI) to evaluate the binning performance on real-world metagenomic data.
Preborn infant gut metagenome (Sharon et al., 2013) with 18 samples (NCBI accession number SRA052203), referred as
Metagenomics of the Chronic Obstructive Pulmonary Disease (COPD) Lung Microbiome (Cameron et al., 2016) with 18 samples (NCBI BioProject number PRJEB9034), referred as
Human metagenome sample from tongue dorsum of a participant from the Deep Whole-Genome Sequencing (WGS) Human Microbiome Project (HMP) clinical samples (Lloyd-Price et al., 2017) with 8 samples (NCBI accession number SRX378791), referred as
Refer to Table 1 for further details of all the datasets.
Further Information of the Datasets
Density of the graph is calculated as (the number of edges)/(the number of vertices).
CAMI, Critical Assessment of Metagenomic Interpretation; CAMI GI, CAMI Urogenital; CAMI UG, CAMI Gastrointestinal; COPD, Chronic Obstructive Pulmonary Disease.
3.1.4. Tools used
We used the popular metagenomic assembler metaSPAdes (Nurk et al., 2017; from SPAdes version 3.15.2 (Bankevich et al., 2012)) to assemble reads into contigs and obtain the assembly graphs. For the datasets containing multiple samples, the contigs and assembly graph were obtained by coassembling the reads from all the samples together. The mean coverage of each contig in each sample was calculated using CoverM (available at https://github.com/wwood/CoverM).
MetaCoAG was benchmarked against the binning tools MaxBin2 (version 2.2.7; Wu et al., 2015), MetaBAT2 (version 2.12.1; Kang et al., 2019), and Vamb (version 3.0.1; Nissen et al., 2021). MaxBin2 uses probabilistic models and an Expectation Maximization (EM) algorithm to iteratively bin the contigs based on their normalized tetranucleotide frequencies and coverage profiles. MetaBAT2 uses normalized tetranucleotide frequency scores and coverage profiles to construct a graph and performs clustering on this graph to bin contigs. Vamb makes use of deep variational autoencoders to encode normalized tetranucleotide frequencies and coverage profiles of contigs and performs clustering on the encoded information. MaxBin2 was run in its default settings, MetaBAT2 with the parameter -m 1500 and Vamb in coassembly mode (for a fair comparison with other tools) with the parameter - -min-fasta 200000 as per authors. The commands used to run these tools can be found in Figure 3.

Commands used to run the different binning tools.
The binning results were evaluated using the tools Assessment of Metagenome BinnERs (AMBER) (Meyer et al., 2018; version 2.0.2), CheckM (Parks et al., 2015; version 1.1.3), and Genome Taxonomy Database-Toolkit (GTDB-Tk) (Chaumeil et al., 2019; version 1.5.0).
3.2. Evaluation metrics
We used Minimap2 (Li, 2018) to map the contigs to the reference genomes and determine their ground truth for the simHC+ dataset as the reference genomes of the underlying species were available. With this ground truth annotation of contigs, we used AMBER (Meyer et al., 2018) to assess the binning results of the simHC+ dataset. We define the AMBER F1-score as follows.
where
For all the datasets, we determined the completeness and contamination of each bin produced by all the binning tools using CheckM (Parks et al., 2015). We define the CheckM F1-score for each bin as follows.
where
Furthermore, considering purity (precision) and completeness based on the CheckM results, we counted the number of high-quality bins (bins with
4. RESULTS AND DISCUSSION
4.1. Benchmarks using simHC+ dataset
We first benchmarked MetaCoAG against two popular contig-binning tools, MaxBin2 (Wu et al., 2015) and MetaBAT2 (Kang et al., 2019), on the simulated dataset
AMBER and CheckM Evaluation Results for the simHC+ Dataset
Bold values are the best evaluation results.
MetaCoAG has recovered bins with a better trade-off between purity and completeness when compared to other binning tools (Fig. 4a). This better trade-off is demonstrated from the best F1-score results produced by MetaCoAG with a median F1-score of 95.69% from AMBER and a median F1-score of 98.48% from CheckM (Fig. 4b and c, respectively, where each point denotes a bin) when compared with other binning tools. Furthermore, MetaCoAG has recovered the highest number of high-quality bins (69 bins) and the lowest number of low-quality bins (13 bins; refer to Table 4).
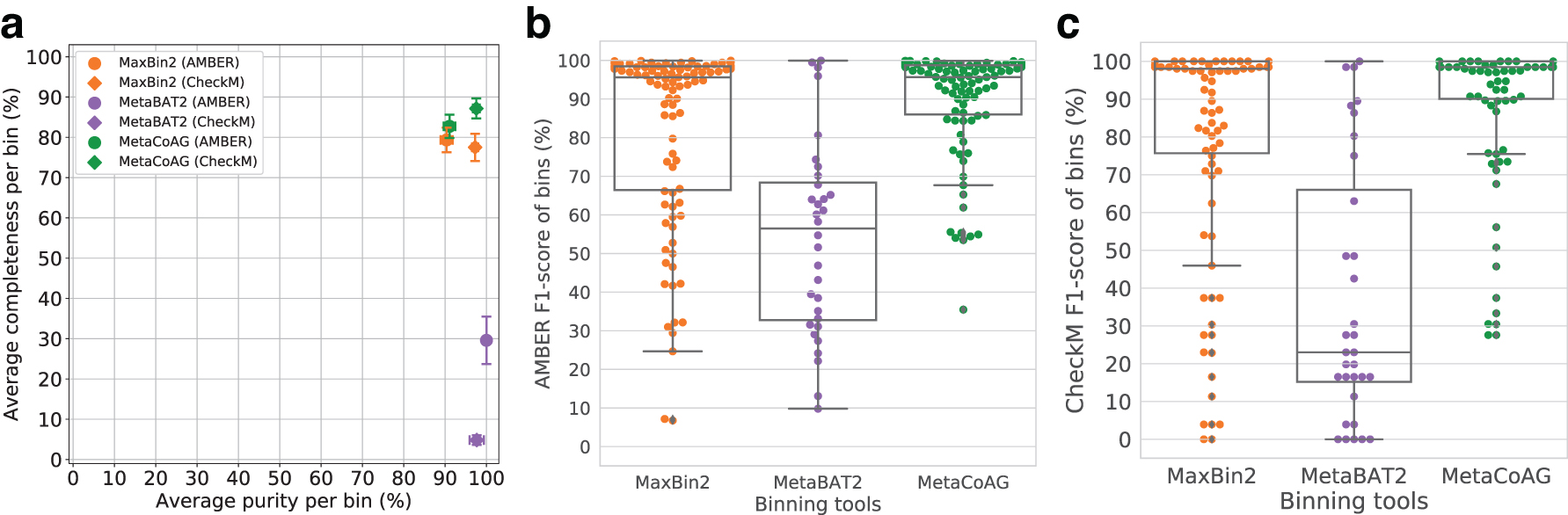
Binning results of the simHC+ dataset:
Many existing binning tools assume that the oligonucleotide composition and coverage are conserved across the genome. Hence, it is challenging for such tools to bin species with high variance in oligonucleotide composition and/or coverage. In Figure 5, we visualize and compare the binning results of MaxBin2 and MetaCoAG
†
against the ground truth for the following species, Pseudomonas putida and Arthrobacter arilaitensis. The species Pseudomonas putida has a high variance in oligonucleotide composition (standard deviation
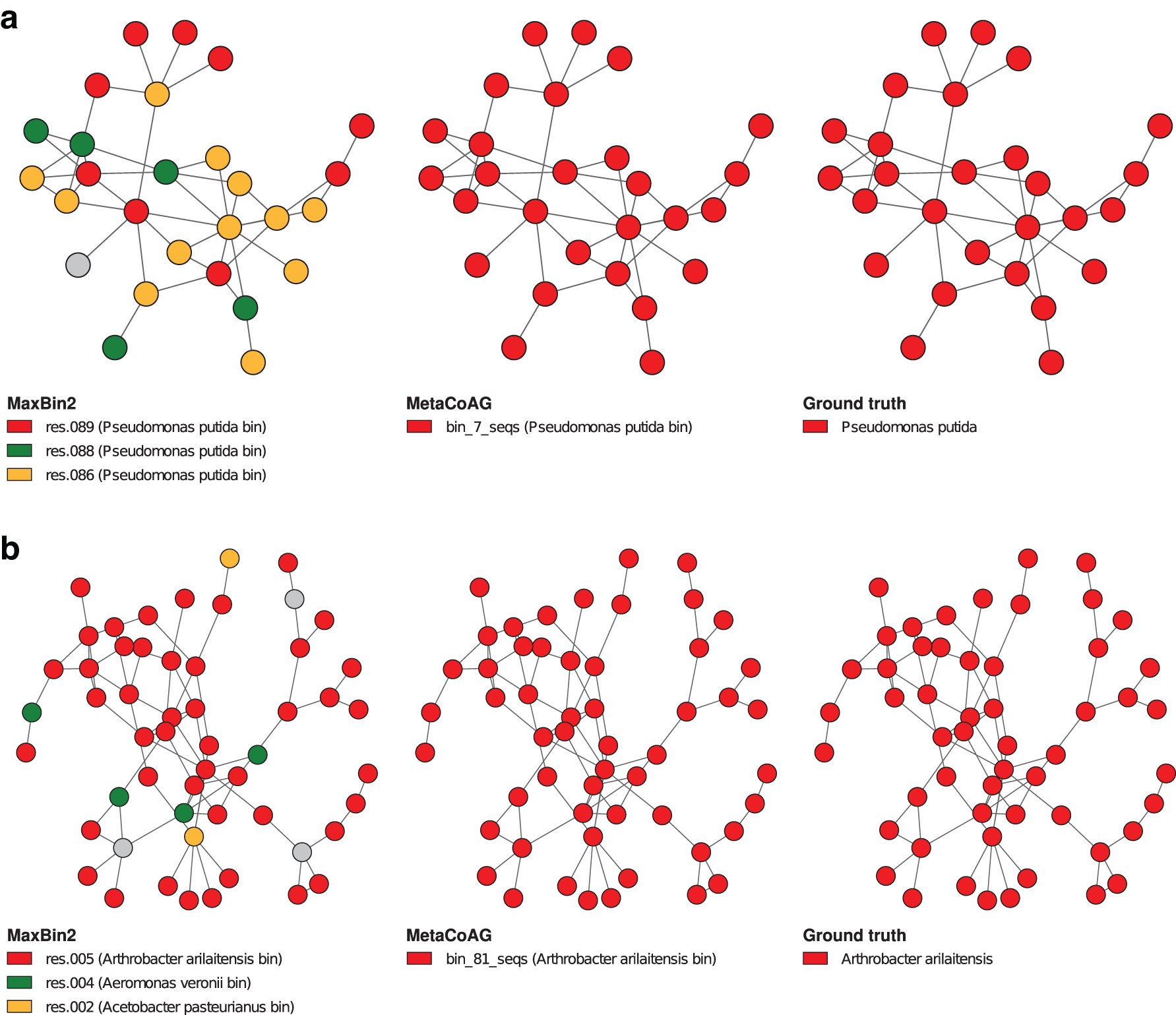
Visualization of the binning results of simHC+ dataset from MaxBin2 and MetaCoAG for a species with
Another major challenge faced by most of the existing metagenomic binning tools is how to accurately separate contigs of species belonging to the same genus, where such species tend to have similar oligonucleotide composition and appear in similar abundances. For example, the three species, S. pneumoniae, S. thermophilus, and S. suis from simHC+ belong to the Streptococcus genus, and they have very similar oligonucleotide composition and coverage values (refer to Fig. 6a). Not surprisingly, contigs from these three species were incorrectly binned by MaxBin2 and even ignored by MataBAT2 because they share similar composition and coverage profiles (refer to Fig. 6b).
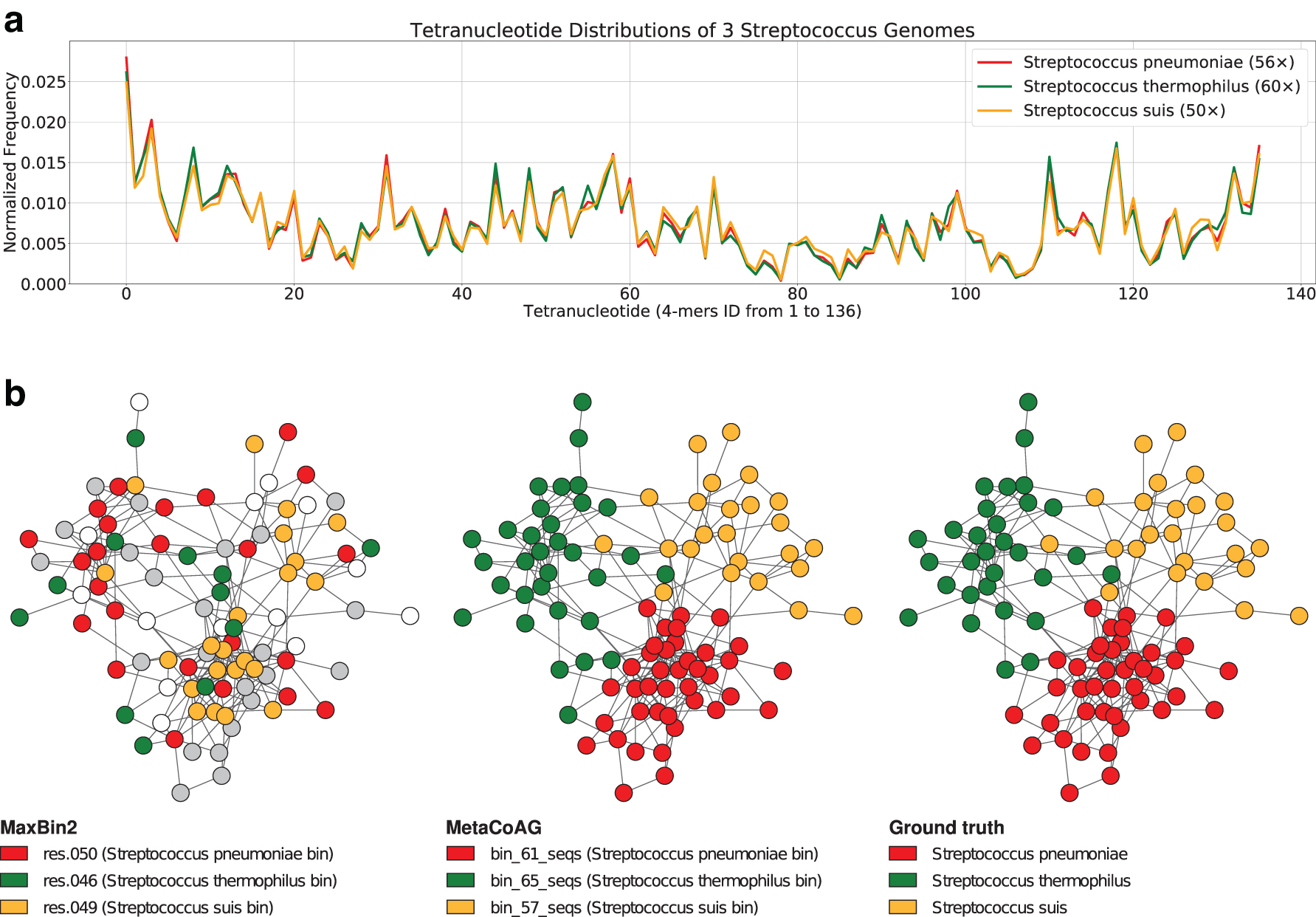
Visualization of the
On the contrary, MetaCoAG was able to accurately bin most of the contigs from these three species because they naturally form three subgraphs in the assembly graph (refer to Fig. 6b), thus improving the F1-scores of S. pneumoniae from 46.51% to 93.40%, S. thermophilus from 49.97% to 95.67%, and S. suis from 72.39% to 95.95%. Figure 6b demonstrates that the use of assembly graph in MetaCoAG can assist in separating species, despite the high similarity in oligonucleotide composition and coverage of certain species. Furthermore, we observed that the assembly graphs help MetaCoAG to bin species with high variance of intraspecies oligonucleotide composition and coverage profiles while most existing tools suffer from the assumption that the oligonucleotide composition and coverage are conserved within the same species.
4.2. Benchmarks using CAMI2 toy human microbiome project datasets
We benchmarked MetaCoAG against MaxBin2 (Wu et al., 2015), MetaBAT2 (Kang et al., 2019), and Vamb (Nissen et al., 2021) on five publicly available datasets from the toy Human Microbiome Project dataset of the second CAMI challenge (Meyer et al., 2022; refer to Table 1 for further details of the CAMI datasets). Multiple samples from each dataset were coassembled together to obtain the final contigs for binning.
We evaluated the binning results of the CAMI datasets using CheckM (Parks et al., 2015) and reported the F1-score of the bins produced by all the binning tools (refer to equation 6). Figure 7a–e shows that overall MetaCoAG has achieved the best binning results among all the binning tools. The overall median F1-scores averaging from all 5 CAMI datasets for MetaCoAG, MaxBin2, MetaBAT2, and Vamb are 86.77%, 75.41%, 1.57%, and 33.30%, respectively. More specifically, MetaCoAG has recovered more complete bins with higher purity and lower contamination when compared to other tools.

Swarm plots with overlaid box plots for the F1-score from CheckM results of the CAMI datasets: (
MetaCoAG produced the highest numbers of high-quality and medium-quality bins combined together for all the CAMI datasets (refer to Table 4). Note that only MaxBin2 outperforms MetaCoAG in terms of the number of high-quality bins just for the CAMI GI dataset. This dataset had a low density in its assembly graph (refer to Table 1 for density of the assembly graph), which prevented MetaCoAG from making full use of the assembly graphs.
4.3. Benchmarks using real datasets
We benchmarked MetaCoAG against MaxBin2 (Wu et al., 2015), MetaBAT2 (Kang et al., 2019) and Vamb (Nissen et al., 2021) on three real metagenomic datasets;
Figure 8 shows that MetaCoAG has also achieved the best binning result in terms of the median F1-score for the real datasets. For the Sharon dataset, MetaCoAG records a median F1-score of 99.24% while the second-best tool (Vamb) has a median F1-score of 83.88%. For the COPD dataset, MetaCoAG records a median F1-score of 75.68% while the second-best tool (MaxBin2) has a median F1-score of 25.13%. For the Deep HMP TD dataset, MetaCoAG records a median F1-score of 76.34% while the second-best tool (MaxBin2) has a median F1-score of 37.40%. Furthermore, MetaCoAG has produced the highest number of high-quality bins for all the real datasets (refer to Table 4).
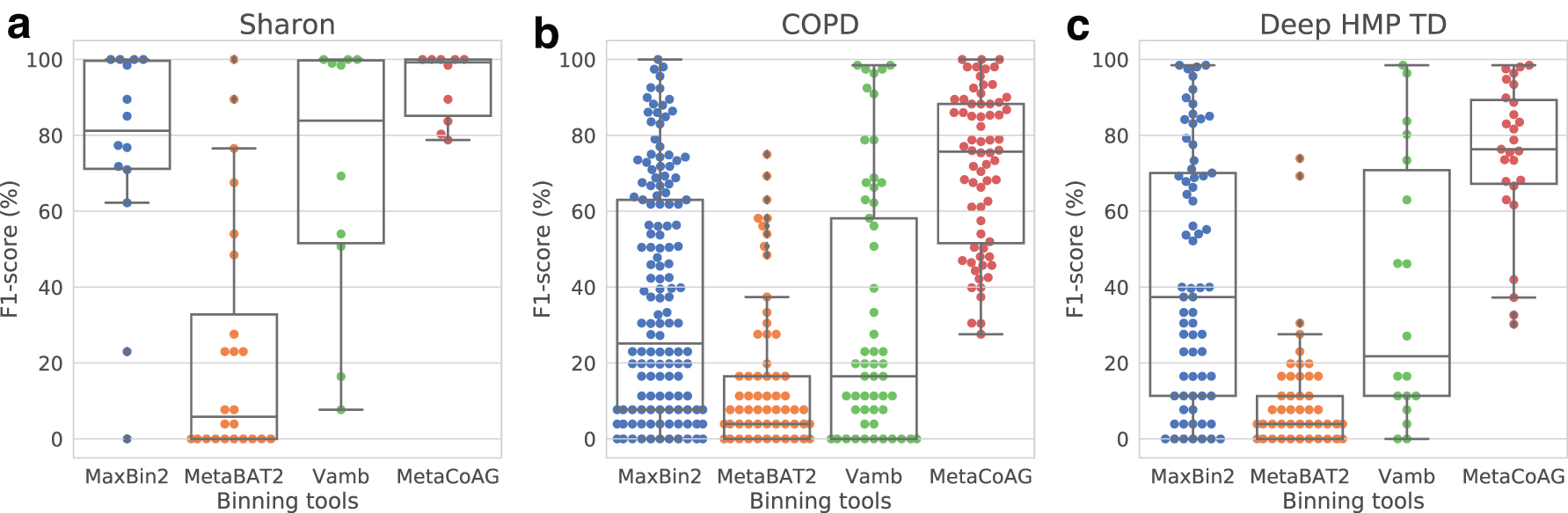
Swarm plots with overlaid box plots for the F1-score from CheckM results of the real datasets: (
We used GTDB-Tk (Chaumeil et al., 2019) to annotate all the high-quality bins produced by the three best-performing tools; MetaCoAG, MaxBin2, and Vamb ‡ for the real datasets. Then we compared the taxonomic annotations (up to the species level) with the analysis results reported by the authors of these datasets as shown in Table 3. According to Table 3, MetaCoAG achieves the best consistency with the original analysis reported by the authors. In the Sharon dataset, the five most abundant species reported according to the authors (Sharon et al., 2013), Staphylococcus epidermidis, Enterococcus faecalis, Cutibacterium avidum, Peptoniphilus lacydonensis, and Staphylococcus aureus, have been successfully identified by all the three binning tools. However, Vamb missed Staphylococcus hominis, which is reported as a rare species in the Sharon dataset (Sharon et al., 2013). Moreover, MetaCoAG is the only tool that is able to recover Leuconostoc citreum, which is also identified as a rare species in the Sharon dataset (Sharon et al., 2013). These results denote the ability of MetaCoAG to recover rare species in real metagenomics samples that are ignored by other binning tools.
GTDB-Tk Annotations of High-Quality Species for the Real Datasets
✓ denotes that the species is present and ✗ denotes that the species is absent in the result/analysis. Bold items match the original analysis, whereas the gray colored items do not match the original analysis.
The species were determined based on the most abundant genera presented.
These species were added to National Center for Biotechnology Information taxonomy in year 2020 (Schoch et al., 2020), which is after the relevant analysis (Cameron et al., 2016; Lloyd-Price et al., 2017).
Vamb, Variational autoencoder for metagenomic binning.
The Number of High-Quality, Medium-Quality, and Low-Quality Bins
Bold values are the best results.
In the COPD dataset, there is a larger discrepancy among MaxBin2, Vamb, and MetaCoAG. Only two species, Peptostreptococcus sp. and SR1 bacterium human oral taxon HOT-345, have been identified by all the three binning tools. SR1 bacterium human oral taxon HOT-345 and Lachnospiraceae bacterium oral taxon 096 have been added to NCBI taxonomy recently (Schoch et al., 2020) and hence are not found in the original analysis (Cameron et al., 2016). Compared to MetaCoAG, MaxBin2 failed to identify three species Prevotella pallens, Prevotella shahii, and Prevotella histicola while Vamb only identified P. pallens under the genus Prevotella. Similarly, Vamb failed to identify two species, Capnocytophaga gingivalis and Capnocytophaga leadbetteri, both of which are identified by MaxBin2 and MetaCoAG. Moreover, the species Anaeroglobus micronuciformis only identified by MaxBin2 was not present in the top 50 genera ranked by abundance in the original analysis (Cameron et al., 2016), which is likely to be a false-positive.
In the Deep HMP TD dataset, the species identified by MetaCoAG show the best consistency with the original analysis (Lloyd-Price et al., 2017), while being the only tool to identify the species from the genus Eubacterium. These results demonstrate that MetaCoAG has been able to recover species in real metagenomics samples that are ignored by other binning tools, as well as recover more species correctly with respect to the original analysis of these real datasets.
4.4. Implementation, running time, and memory usage
The source code of MetaCoAG was implemented using Python 3.7.4. FragGeneScan version 1.31 and HMMER version 3.3.2 were used in MetaCoAG to scan for single-copy marker genes in the contigs. MetaCoAG allows the users to input custom marker sets (e.g., for fungus and protists) instead of using the default bacterial markers.
Table 5 denotes the running times and memory usage of all the binning tools for all the datasets. All the binning tools were run on a Linux system with Ubuntu 18.04.1 LTS, 16 GB memory and Intel(R) Core(TM) i7-7700 CPU @ 3.60 GHz with 4 CPU cores. All the binning tools were run using 8 threads. MetaCoAG has completed running under 1 hour and 5 minutes for all the datasets.
Running Time and Memory Usage of the Different Binning Tools for All the Datasets
h, hours; m, minutes; s, seconds.
5. DISCUSSION AND CONCLUSION
High-throughput sequencing and de novo assembly of metagenomes, together with metagenomic binning methods have paved the way to characterize different microbial communities and construct draft microbial genomes of previously uncharacterized micro-organisms via MAGs. Most of the existing metagenomic contig-binning tools do not make use of the valuable connectivity information found in assembly graphs, from which the contigs are derived. Furthermore, existing tools do not make full use of multiple single-copy marker genes during the entire binning process.
We have developed MetaCoAG, an automated tool for binning metagenomic contigs using assembly graphs along with composition and coverage information of contigs. The usage of connectivity information from assembly graphs makes the binning process of MetaCoAG robust against similar interspecies oligonucleotide composition and coverage (among species within the same genus) as well as high variance of intraspecies oligonucleotide composition and coverage (within the same species). From our experimental results, we show that MetaCoAG achieves the best binning performance for both simulated and real datasets when compared to state-of-the-art tools, especially in terms of bin quality. However, problems in assembly such as misassemblies can be challenging to handle and require further investigation.
MetaCoAG can be easily extended to work with other popular metagenomic assemblers such as MEGAHIT (Li et al., 2015) and metaFlye (Kolmogorov et al., 2020). In the future, we plan to extend MetaCoAG to support overlapped binning (Mallawaarachchi et al., 2020b, 2021; i.e., identifying contigs that may belong to multiple species) and multisampled binning (Nissen et al., 2021; i.e., integrating contigs from assemblies across multiple samples instead of performing coassembly). Furthermore, we plan to incorporate MetaCoAG with metagenomic analysis pipelines that may lead to more efficient and accurate analysis for metagenomic datasets.
DATA AND CODE AVAILABILITY
All the CAMI and real datasets containing raw sequencing data used for this study are publicly available from their respective studies. The CAMI2 Toy Human Microbiome Project datasets were downloaded from https://data.cami-challenge.org/participate from the second CAMI Toy Human Microbiome Project Dataset. The Sharon dataset was downloaded from NCBI with BioProject number PRJNA60717 and accession number SRA052203. The COPD dataset was downloaded from NCBI with BioProject number PRJEB9034, and the NCBI accession numbers of the runs used in this study are ERR970477, ERR970476, ERR970475, ERR970474, ERR970473, ERR970472, ERR970471, ERR970470, ERR970407, ERR970406, ERR970405, ERR970404 ERR970403, ERR970402, ERR970401, ERR970400, ERR970399, and ERR970398. The Deep HMP TD dataset was downloaded from NCBI with BioProject number PRJNA48479 and the NCBI accession numbers of the runs used in this study are SRR1031078, SRR1031179, SRR1031181, SRR1031229, SRR1031267, SRR1031290, SRR1031684, and SRR1031924.
All the assembled data and results from all the binning tools, including the source data for Figures 4–8 and Table 3, are available on figshare at https://figshare.com/projects/MetaCoAG/121014. The sequencing data and assembly files for the simulated simHC+ dataset can be found at https://cloudstor.aarnet.edu.au/plus/s/h44eWFUhCWwGQl7.
The code of MetaCoAG is freely available on GitHub under the GPL-3.0 license and can be found at https://github.com/metagentools/MetaCoAG. All analyses in this study were performed using MetaCoAG v.1.0 with default parameters. MetaCoAG is also available as a conda package on bioconda at https://anaconda.org/bioconda/metacoag.
Footnotes
ACKNOWLEDGMENTS
This research was undertaken with the assistance of resources and services from the National Computational Infrastructure (NCI), which is supported by the Australian Government.
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
No funding was received for this article.