
Abstract
TRIMER, Transcription Regulation Integrated with MEtabolic Regulation, is a genome-scale modeling pipeline targeting at metabolic engineering applications. Using TRIMER, regulated metabolic reactions can be effectively predicted by integrative modeling of metabolic reactions with a transcription factor-gene regulatory network (TRN), which is modeled through a Bayesian network (BN). In this article, we focus on sensitivity analysis of metabolic flux prediction for uncertainty quantification of BN structures for TRN modeling in TRIMER. We propose a computational strategy to construct the uncertainty class of TRN models based on the inferred regulatory order uncertainty given transcriptomic expression data. With that, we analyze the prediction sensitivity of the TRIMER pipeline for the metabolite yields of interest. The obtained sensitivity analyses can guide optimal experimental design (OED) to help acquire new data that can enhance TRN modeling and achieve specific metabolic engineering objectives, including metabolite yield alterations. We have performed small- and large-scale simulated experiments, demonstrating the effectiveness of our developed sensitivity analysis strategy for BN structure learning to quantify the edge importance in terms of metabolic flux prediction uncertainty reduction and its potential to effectively guide OED.
INTRODUCTION
Optimal experimental design (OED) and control for complex biological systems have significant impact on developing new computational strategies in systems and synthetic biology (Balsa-Canto et al, 2021; Zhao et al, 2020) for targeted biochemical overproduction that may benefit human society, for example, in different energy-related and pharmaceutical applications (Barrett et al, 2006; Bro et al, 2006; Esvelt and Wang, 2013; Haro and de Lorenzo, 2001; Lü et al, 2011; Luengo et al, 2003; Ohta et al, 1991). In particular, metabolic engineering, which genetically redesigns microbial strains by gene or reaction knockouts, aims to optimize corresponding biological processes with respect to the desired engineering objective(s).
Owing to the demanding experimental cost and time to test different microbial strains in vivo, computational methods have been developed for in silico prediction of useful knockout strategies for beneficial mutants. Mathematical models to systematically analyze genome-scale metabolic reaction networks have been developed to derive optimal intervention strategies to achieve the desired metabolic reaction fluxes, the turnover rates of the molecules through the corresponding metabolic pathways (Edwards and Palsson, 2000; Segre et al, 2002; Varma and Palsson, 1994).
However, many existing computational methods to obtain genetically engineered strains are based on genome-scale analysis at steady states assuming the static network models (Apaydin et al, 2017; Apaydin et al, 2016; Burgard et al, 2003; Edwards and Palsson, 2000; Ren et al, 2013; Segre et al, 2002; Shlomi et al, 2005; Varma and Palsson, 1994). Recent efforts have integrated genetic regulatory relationships involving transcriptional factors (TFs) that may regulate metabolic reactions to achieve more accurate and robust prediction of target metabolic behaviors under different conditions or contexts (Chandrasekaran and Price, 2010; Covert and Palsson, 2003; Covert et al, 2008; Machado and Herrgård, 2014; Motamedian et al, 2017; Reed, 2017; Shlomi et al, 2007; Yu and Blair, 2019).
To generalize these integrated hybrid models, we have developed Transcription Regulation Integrated with MEtabolic Regulation (TRIMER) (Niu et al, 2021) as a modeling pipeline targeting at metabolic engineering applications. Using TRIMER, regulated metabolic reactions can be effectively predicted by integrative modeling of metabolic reactions with a TF-gene regulatory network (TRN), where the TRN is modeled through a Bayesian network (BN) inferred from transcriptomic expression data. We have demonstrated promising metabolic flux prediction performances in both simulated and real-world microbial mutant design applications considering transcription regulation in genome-scale metabolic prediction (Niu et al, 2022; Niu et al, 2021).
Although all the existing efforts have demonstrated valid performances in selected model organisms with abundant data and careful manual curation, there is not much investigation on how model uncertainty, due to incomplete system knowledge and/or limited training data, may affect metabolic predictions. To the best of our knowledge, most of the existing works assume that the trained models are deterministic without considering potential model uncertainty. Compared with the traditional ways where model uncertainty is often represented by how well models fit the training data, we propose to analyze how model uncertainty affects the metabolic engineering performance directly.
In particular, we analyze metabolic flux prediction sensitivity with respect to the uncertainty class of BN structures for TRN modeling as one possible way of uncertainty quantification. Through a mathematical programming formulation of BN topological ordering, we construct uncertainty classes of BN models for TRN and analyze the metabolic prediction sensitivity of our TRIMER modeling pipeline. We evaluate the sensitivity of the TRIMER pipeline by comparing the ground-truth metabolite yield alterations with TF knockout mutations based on the BN uncertainty classes with the network edge space ranked by topological ordering.
To be specific, we simulate both gene expression and metabolic flux data from a predefined ground-truth TRN-regulated metabolic network model, infer models from the simulated data, construct uncertainty classes, and then check the prediction sensitivity by checking the correlation of the predicted and ground-truth metabolite yields. The obtained sensitivity analyses can provide useful guidance for model learning, calibration, and OED for metabolic engineering, allowing biologists to better understand metabolism under perturbation and to take advantage of high-throughput genetic engineering for desired microbial strains with reduced cost.
BACKGROUND
In Niu et al (2022, 2021), we have developed an integrated regulatory-metabolic hybrid network model and genome-scale metabolic analysis pipeline, TRIMER. TRIMER enables condition-dependent genome-scale metabolic behavior modeling and provides in silico predictions of metabolic engineering tasks, such as knockout phenotype and knockout flux predictions. As a hybrid network model, TRIMER has a metabolic network module that predicts metabolic fluxes based on the classic flux balance analysis (FBA) framework (Covert and Palsson, 2003; Edwards and Palsson, 2000; Lewis et al, 2010; Palsson, 2015; Varma and Palsson, 1994).
This well-known technique adopts linear programming with flux constraints introduced by metabolic regulation rules (i.e., a metabolic network) of a given organism, to model genome-scale steady-state metabolic status. For accurate and robust prediction of metabolic behaviors under different conditions, regulatory relationships, in the corresponding TF regulatory network (TRN) between TFs and their target genes, are integrated with the FBA formulation as additional transcriptional constraints over fluxes (Apaydin et al, 2017; Covert et al, 2008; Jensen et al, 2011; Ren et al, 2013; Shlomi et al, 2007; Shlomi et al, 2005).
Motivated by another genome-scale framework PROM (probabilistic regulation of metabolism) (Chandrasekaran and Price, 2010), TRIMER adopts a TRN module that integrates the a priori known TF-gene interaction annotations and available gene/transcriptomic expression profiles to learn a corresponding BN for probabilistically modeling the TRN. The conditional probabilities
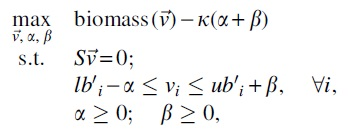
where S is the stoichiometric matrix deduced from the given metabolic network,
In practice, the BN is learned from a gene expression data set with a limited number of data points, giving rise to potential uncertainty in BN structures, the estimated conditional probabilities
Although TRIMER is motivated by PROM, the adopted flux bounds in PROM are based on
MATERIALS AND METHODS
In this section, we first provide a brief overview of our proposed BN structure sensitivity analysis strategy, followed by detailed descriptions of all the steps of the procedure.
Overview: sensitivity analysis of TRIMER
Sensitivity analysis of TRIMER is achieved by evaluating metabolic flux prediction performances based on the uncertainty classes of BNs modeling TRN. In particular, we focus on the uncertainty of BN structures that directly impacts metabolic flux prediction. To construct an uncertainty class, the key idea is to first infer the BN topological ordering learned from gene expression data. We then grow various BN structures from the inferred node ordering to construct uncertainty classes at different perturbation levels.
By statistical analysis of metabolic predictions with different uncertainty classes, we can have better understanding on how TRN modeling may affect the final metabolic predictions in the TRIMER hybrid model. With such sensitivity analyses as a way of the BN model uncertainty quantification, practical guidance can be obtained inform researchers for active learning and calibrating the modular components in TRIMER through optimal experiment design, for example, defining the iterative structure updating policies for BN to improve metabolic prediction.
To develop such a sensitivity analysis capability in TRIMER, we first implement a BN topological order search algorithm to infer the network node ordering of BNs given transcriptomic expression data and the search space of BN edges. The topological ordering of BNs determines the parenthood of nodes: an ancestor node must be of higher order than its descendant nodes. Therefore, given node parent sets and a topological ordering of interest, the global optimal consistent structure can be obtained. To measure the uncertainty of the corresponding topological ordering, ordering samples are obtained by bootstrapping the adopted order search algorithms (Friedman et al, 1999). Accordingly, the statistics of bootstrapped ordering samples can be computed to help deriving the probability distributions and thereafter uncertainty quantification by constructing BN uncertainty classes.
The next question is how we may capture the potential model uncertainty from bootstrapped BN topological orderings and derive a rigorous mathematical framework to construct the classes of uncertain BN models for our proposed metabolic prediction sensitivity analysis. Following Xiao et al (2018), we adopt a mathematical programming formulation to rank the pairwise ordering of nodes or directed edges in the BN modeling TF-gene regulatory relationships. The formulation aims at identifying the critical regulatory relationships for which reducing the ordering uncertainty can help significantly improve the BN model fitting to the given training data set.
Solving this problem, all valid edges in the given edge space for the BN are ranked by their contributions to the uncertainty reduction, where the uncertainty is captured by the corresponding covariance matrix of ordering scores computed from bootstrapped ordering samples. We then construct the BN uncertainty class of different sizes, where edges with higher rankings are more likely to be sampled to form the allowed edge space to derive the best BN structure consistent with the topological ordering. Finally, transcription-regulated genome-scale metabolic predictions with the BNs in the corresponding uncertainty class can be done following the TRIMER pipeline to investigate prediction sensitivity. The details of the TRIMER analysis pipeline can be referred to Niu et al (2022).
In summary, our proposed strategy mainly comprises three steps: (1) BN topological ordering, (2) uncertainty class construction, and (3) metabolic prediction sensitivity analysis. Figure 1 provides a high-level overview of the strategy, depicting the main workflow.
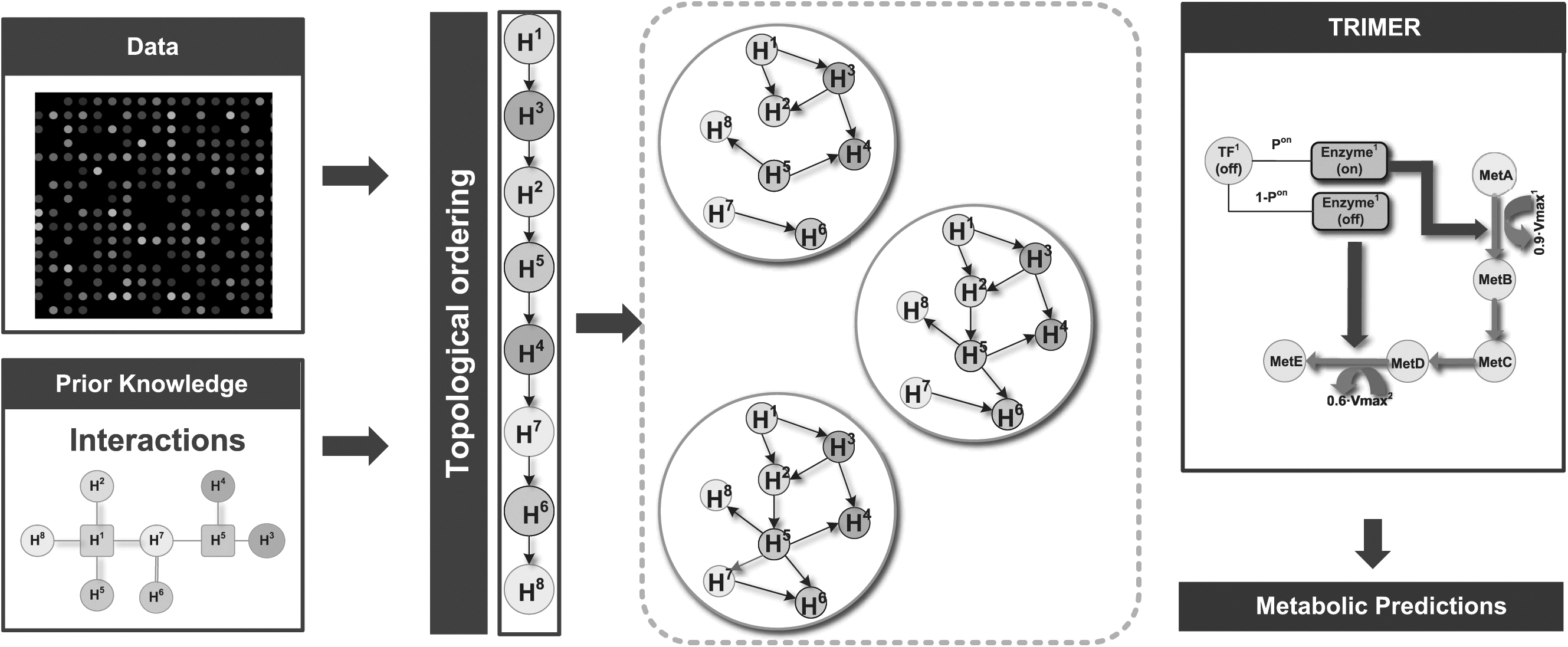
Schematic illustration of the proposed sensitivity analysis workflow: (1) gene expression data with the prior knowledge on regulatory interactions are used to infer the topological orderings of nodes in BNs. (2) The uncertainty class of BNs is constructed based on the uncertainty of the topological ordering due to incomplete knowledge and/or limited data. (3) TRIMER pipeline is used with the uncertainty class to analyze the metabolic flux prediction sensitivity. TRIMER, Transcription Regulation Integrated with MEtabolic Regulation.
In the TRIMER pipeline (Niu et al, 2021), BN structure learning is by fitting the given gene expression profiles D for the corresponding gene set

where

The best swapping operation is selected among all
Order initialization
Initialization is crucial to guarantee satisfactory performance of heuristic search as the exact optimality is not ensured due to both the constrained search space and the greedy nature. In this article, the TF-gene interaction list is not just used to help define the edge space but also as the prior knowledge of node ordering. For example, the ordering of the TF nodes cannot be lower than its regulated gene nodes. Otherwise, the corresponding TFs cannot be the parent nodes of its regulated genes in BNs. In the following, we will introduce an order initialization algorithm through extracting the node ordering prior knowledge from a given TF-gene interaction list.
In the extreme case, the edge space E defined by a given interaction list corresponds to a directed acyclic graph (DAG), for which the optimal BN topological ordering is just any ordering consistent with the graph structure. However, in practice, the edge space almost always corresponds to a directed graph with cycles. Although the optimal node ordering cannot be directly read off from the graph structure, it is still possible to obtain prior knowledge about it. For a directed graph
A graph of C can be denoted as
To obtain an appropriate initial node ordering
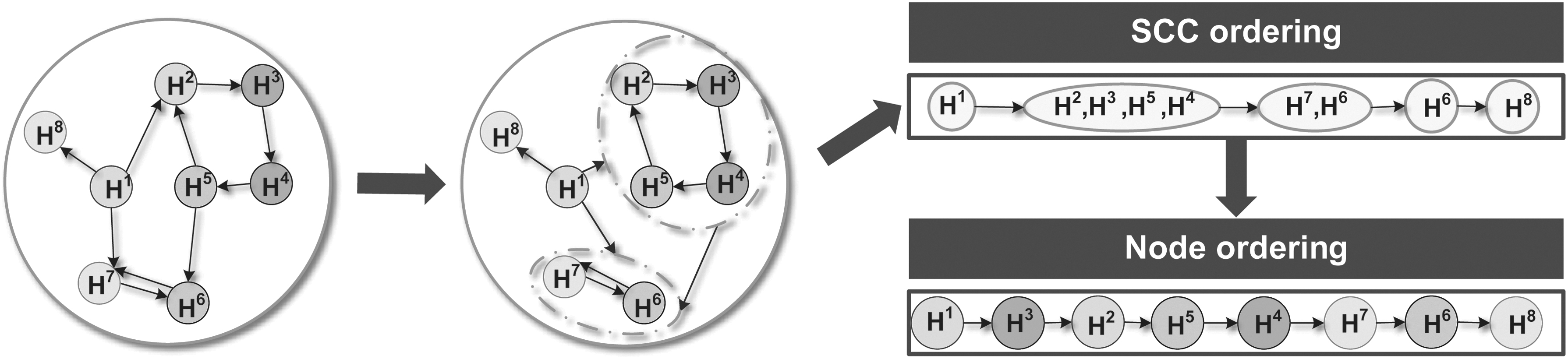
Illustration of the order initialization workflow. First, the DAG of SCCs is identified from the given directed graph; then a node-wise ordering consistent with the SCC ordering is randomly selected. DAG, directed acyclic graph; SCC, strongly connected component.

To quantify the uncertainty of topological ordering, we use a multivariate Gaussian random vector
To establish the relationship between node-wise orders and edges, we associate edge
In cases when only a subset of E is considered, a binary matrix
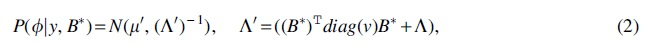
where

In the formulation aforementioned,  to guarantee the convexity of the SDP. While this relaxation leads to potential interpretation ambiguity of the solution as the number of nonzero elements in v can be higher than
to guarantee the convexity of the SDP. While this relaxation leads to potential interpretation ambiguity of the solution as the number of nonzero elements in v can be higher than
where i denotes the index of an edge in E and
As described in Niu et al (2021), TRIMER can serve as a simulator of gene expression and metabolic flux data. Given a ground-truth BN model for TRN with appropriate conditional probability tables for each node in the BN, gene expression data sets can be simulated by drawing samples from the distribution described by the BN. Moreover, conditional probabilities with respect to TFs and target genes can be inferred from the BN and used for constructing regulatory flux constraints for genome-scale metabolic predictions when they are integrated with the available metabolic reaction network model of the organism under study. Adding these new constraints into the corresponding FBA formulation for the metabolic network, condition-dependent metabolic states of the organism can be simulated and treated as the ground truth.
For sensitivity analysis, we simulate gene expression data for BN uncertainty class construction and ground-truth metabolic fluxes to estimate the biomass for different TF-knockout Escherichia coli strains as described in Niu et al (2021). We investigate the model uncertainty by computing the corresponding Pearson correlation coefficients (PCCs) between ground-truth biomass fluxes and the predicted fluxes by TRIMER based on BN uncertainty classes, of which the BN structures may significantly deviate from the ground-truth model that simulates the data.
RESULTS
In this section, we present the experimental results based on two simulated data sets to demonstrate the effectiveness of proposed regulatory order-based sensitivity analysis strategy for TRIMER.
Sensitivity analysis results
In our experiments, simulated ground-truth TRIMER models are used to generate gene expression data as well as metabolic flux data (Niu et al, 2021), which are used as the training data sets for BN learning and the ground-truth metabolic flux predictions for sensitivity analysis and performance evaluation. We focus on biomass prediction under multiple TF-knockouts while the proposed methods can be used for other metabolite yield predictions based on the problems of interest.
We here use the two same simulated TRIMER models for E. coli with iAF1260 (King et al, 2016) as their genome-scale metabolic network model as described in Niu et al (2021). One model is based on a small-scale TRN and the other is based on a large genome-scale TRN as detailed in the following subsections. More detailed descriptions on the TRIMER models, data sources, software requirements, hardware setups, as well as run-time statistics of each TRIMER component can be found in Niu et al (2022, 2021). All the reported experiments are implemented on a PC with Intel i7 processor and 16GB RAM.
Small-scale model sensitivity analysis
For the small-scale model, its corresponding TRN contains 50 nodes (12 TFs and 38 regulated genes) with 118 randomly generated edges. It is assumed that regulated genes cannot be the parents of TFs in the BN model. The ground-truth metabolic fluxes in this model are simulated for all the 12 TF single-knockout conditions. Besides, we generate a gene expression data set of 1000 samples from the ground-truth BN model. In light of experimental results reported in Niu et al (2021), a data set of 1000 training samples is believed to be adequate to guarantee reasonable order learning to achieve desired metabolic flux prediction performance.
Then we bootstrap the Tabu order-based search for 100 times over the data set obtaining 100 ordering samples, where one time running of Tabu order-based search typically takes 5–8 minutes. The covariance matrix of ordering scores based on boostrapped ordering samples is shown in Figure 3a. As shown in the figure, the variances corresponding to TFs with indices from 1 to 12 are much smaller than the ones for regulated genes indexed from 13 to 50. This is reasonable as the regulated genes are mostly downstream and their orderings can be more uncertain compared with TFs.
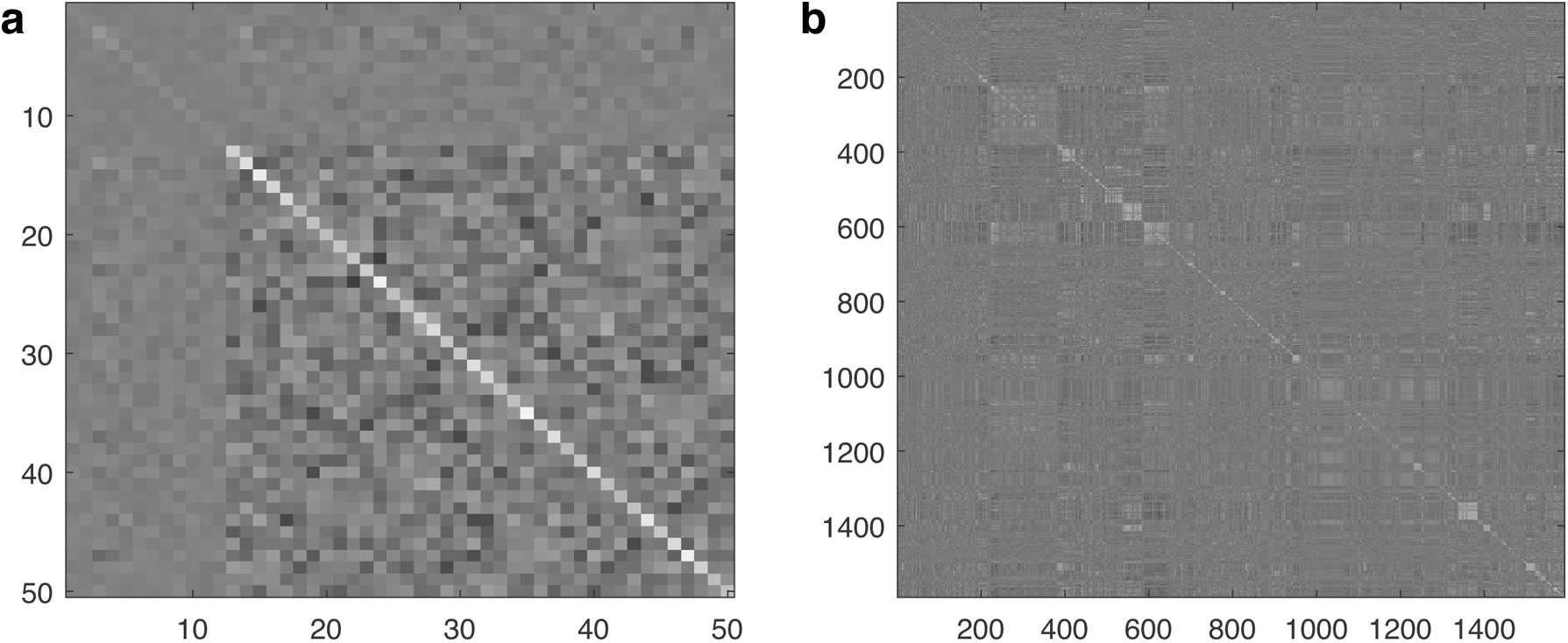
Estimated covariance matrices of topological ordering scores based on bootstrapped samples for
Next, edges in the predefined edge space based on the available prior knowledge for the TRN are ranked by their contribution to uncertainty reduction during the ordering posterior covariance updates through the mathematical programming formulation detailed in Materials and Methods section. As the edge space is relatively small in this small-scale example, the edge ranking can be completed within a few seconds. Ten BN uncertainty classes, each of which contains 10 graph structures consistent with the ordering numerical mean, are constructed from sampled edge sets of sizes ranging from
Finally, the TRIMER model with constructed BN uncertainty classes is built and biomass predictions are made following the TRIMER pipeline. In our experiments, we calculate PCCs between predictions and simulated ground-truth fluxes to evaluate TRIMER's performance under a specific BN configuration in the uncertainty class. We have also performed t-tests between the PCC values of metabolic flux predictions obtained based on the uncertainty class covering the whole edges space and the corresponding PCC values from the other uncertainty classes. The corresponding p-values are calculated to show the statistical significance of performance change. The final metabolic prediction performance by TRIMER with different BN uncertainty classes is illustrated in Figure 4a for this experiment.

Box plots of PCC between the metabolic prediction by TRIMER with constructed BN uncertainty classes and the ground truth. Results are shown for
Increasing the size of edge sample sets from 10% to the whole edge space corresponds to decreasing the uncertainty of BN classes by extending the edge space. As the edge space is extended by the top regulatory relationships in terms of BN topological ordering uncertainty reduction, BN classes of high uncertainty can still maintain essential edges.
To further evaluate the prediction sensitivity of BN models for the small-scale TRIMER model, we conduct an experiment to investigate how its performance varies by changing the size of training gene expression data sets to construct the BN uncertainty classes. We have generated five expression data sets with sizes ranging from 200 to 1000. For each data set, we evaluate the performance of TRIMER with the uncertainty classes constructed from the corresponding sampled edge sets whose sizes are fixed to the half of the complete edge space. The experimental results are depicted in Figure 5, where we provide the t-test p-values based on the corresponding PCC values between the last condition and each of the other conditions.

Box plots of PCC between the metabolic prediction by TRIMER and the ground truth. Results are shown for uncertainty classes constructed based on simulated gene expression data sets of different sizes. As statistical supports, the p-values of the t-tests between the PCCs in the last condition and each PCC set of the other conditions are also shown in the plot to illustrate the statistical significance of the corresponding performance change, with p-values
Overall, with the increasing training samples for BN learning, both the average prediction accuracy, measured by PCC, and sensitivity, shown in the quantile bar plot, improve in general. The prediction performance may not improve much in average when we have >400 training samples. However, when investigating prediction sensitivity to the inherent model uncertainty with our regulatory order-based uncertainty class construction, our results indicate that more training samples may need to guarantee the desired robust predictions. In our experiment, we observe that we require 1000 gene expression samples to achieve accurate and stable predictions.
We conduct another experiment to further investigate how flux predictions may be affected by the noise in gene expression data for BN training. We randomly flip the corresponding ON/OFF states of 10%, 20%, …, 50% genes in a simulated data set of 1000 samples, resulting in five perturbed gene expression data sets at different noise levels. For each data set, we evaluate the performance of TRIMER with the uncertainty classes constructed as previously described based on topological ordering derived from the corresponding perturbed gene expression data. For this experiment, we fix the sampled edge set size to be the half of the complete edge space.
The experimental results are shown in Figure 6, where t-tests are performed between the uncertainty class constructed from nonperturbed data set and each of the other uncertainty classes from data sets perturbed at different levels. From the plot, the performance declines significantly when >
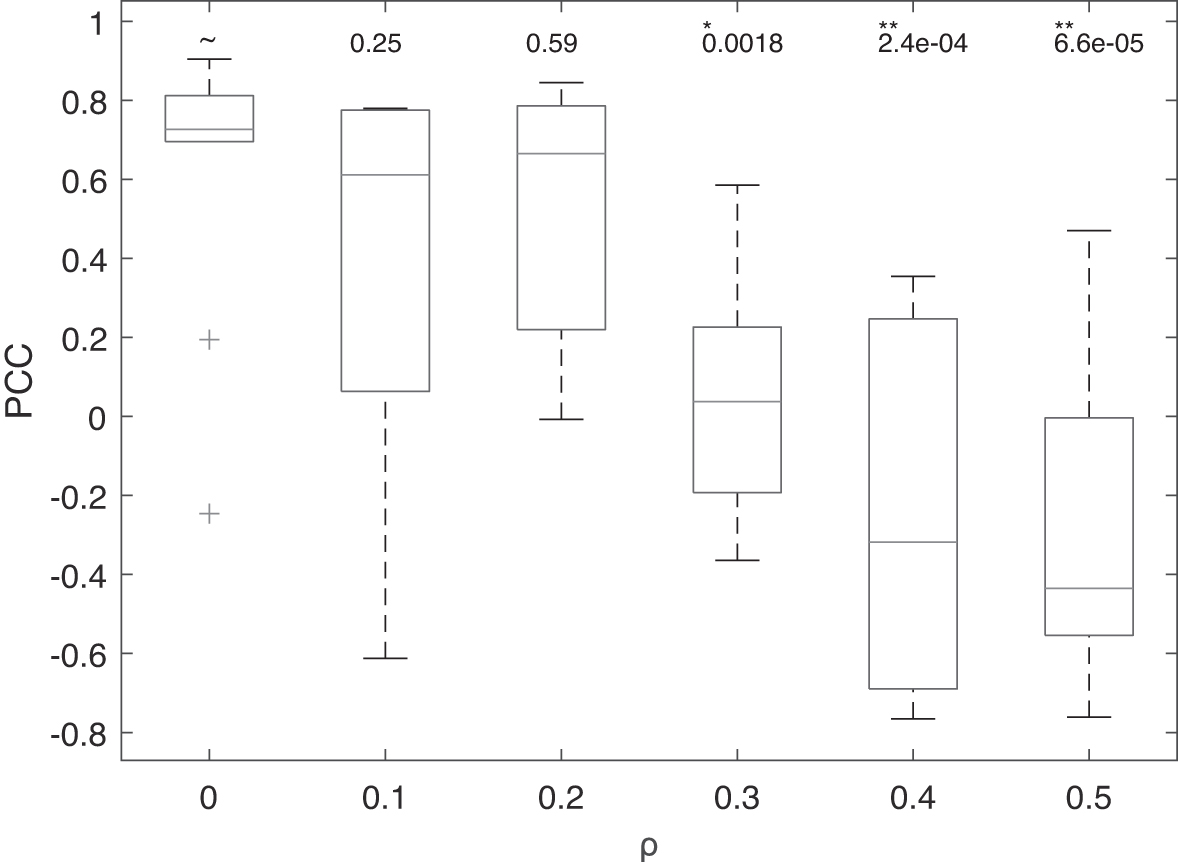
Box plots of PCC between the metabolic flux predictions by TRIMER and the ground truth. Results are shown for the uncertainty classes constructed based on gene expression data perturbed at different levels
All these experiments with this small-scale ground-truth model have demonstrated that our topological ordering based sensitivity analysis strategy can appropriately identify important edges that directly impact our objective of reliably predicting metabolic fluxes in the TRIMER pipeline. When needed, it can help OED in the TRIMER pipeline, where BN can be calibrated and updated iteratively based on the proposed sensitivity analysis. On the contrary, blind random edge sampling may not give rise to an informative performance plot.
For the large-scale model, we consider a genome-scale TRN with 1509 edges randomly selected from the edge space containing 3704 edges in the annotated interaction list for E. coli in EcoMAC (Carrera et al, 2014). When constructing the BN uncertainty classes in this experiment, we only focus on an edge subspace comprised of 1533 edges, which connect genes directly regulating the reactions involving biomass production in the iAF1260 metabolic network model. We obtain 100 ordering samples by bootstrapping the order search over the simulated gene expression data set of 1000 samples, similarly as described in the small-scale model experiment.
In this set of experiments, the cyclic graph deduced from the interaction list is close to an acyclic graph with only 20 SCCs containing more than two nodes, for which one time bootstrapping of Tabu order-based search can be completed within 1–2 minutes. The corresponding covariance matrix is shown in Figure 3b. To construct the BN uncertainty classes, we first fix the BN structure for the genes that are not associated with biomass-related reactions to the optimal structure consistent with the derived mean topological ordering. The run-time of edge ranking for this larger graph is ∼10 minutes. We then sample different sizes of edge sets from the focused edge subspace. The corresponding BN edge space for the uncertainty classes also grows from containing
We observe similar trends as in the small-scale model sensitivity analysis. In general, both metabolic prediction and sensitivity performances improve with the growing edge space. Owing to the integration of the EcoMAC interaction list as prior knowledge for defining the BN edge space, we can achieve satisfactory prediction performance, PCC
CONCLUSION
From our experimental results, it can be observed that when the BN models deviate more from the optimal BN by missing highly ranked critical edges, the prediction accuracy, measured by correlation between ground-truth biomass fluxes and predicted fluxes of the perturbed BN models, does decrease. More critically, when constructing such uncertainty model classes and investigating prediction sensitivity, our results indicate that we may need better prior knowledge and more training data to achieve both accurate and stable predictions. Our sensitivity analyses also indicate that reliable uncertainty quantification may require more data. Although many existing works have reported valid performances in selected experiments, there may still be potential overfitting risks with predictions not easy to generalize when having slightly perturbed systems.
Our topological ordering-based sensitivity analysis also helps identify the set of edges, for which the corresponding uncertainty reduction can significantly help model prediction and improve sensitivity. Such a capability can lead to new uncertainty quantification formulations, which may enable OED strategies for active model learning (Zhao et al, 2021a; Zhao et al, 2021b; Zhao et al, 2021c) and more robust intervention strategies in metabolic engineering, which we leave for future research.
Footnotes
AUTHORs' CONTRIBUTIONS
Methodology, investigation, software, writing—original draft, and writing—review and editing by P.N. Investigation, validation, and writing—review and editing by M.J.S. Conceptualization, methodology, and writing—review and editing by S.H. Conceptualization, methodology, investigation, formal analysis, writing—review and editing, and funding acquisition by B.-J.Y. Conceptualization, investigation, formal analysis, writing—review and editing, and funding acquisition by E.R.D. Conceptualization, investigation, formal analysis, writing—review and editing, resources, and funding acquisition by F.J.A. Conceptualization, investigation, validation, writing—review and editing, funding acquisition, resources, and supervision by I.B. Conceptualization, methodology, investigation, formal analysis, writing—original draft, writing—review and editing, funding acquisition, resources, and supervision by X.Q.
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
P.N. and X.Q. are partially supported by the National Science Foundation under grants CCF-1553281. This study has been supported by the DOE Joint Genome Institute (![]() ) by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research, through contract DE-AC02-05CH11231 between Lawrence Berkeley National Laboratory and the U.S. Department of Energy. This presented material is based upon the study supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under contract number DE-0012704. Publication fees are supported by the U.S. Department of Energy, Office of Science, RadBio program, under Award KP1601011/FWP CC121.
) by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research, through contract DE-AC02-05CH11231 between Lawrence Berkeley National Laboratory and the U.S. Department of Energy. This presented material is based upon the study supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under contract number DE-0012704. Publication fees are supported by the U.S. Department of Energy, Office of Science, RadBio program, under Award KP1601011/FWP CC121.