
Abstract
Despite the recent surge of viral metagenomic studies, recovering complete virus/phage genomes from metagenomic data is still extremely difficult and most viral contigs generated from de novo assembly programs are highly fragmented, posing serious challenges to downstream analysis and inference. In this study, we develop FastViromeExplorer (FVE)-novel, a computational pipeline for reconstructing complete or near-complete viral draft genomes from metagenomic data. The FVE-novel deploys FVE to efficiently map metagenomic reads to viral reference genomes, performs de novo assembly of the mapped reads to generate contigs, and extends the contigs through iterative assembly to produce final viral scaffolds. We applied FVE-novel to an ocean metagenomic sample and obtained 268 viral scaffolds that potentially come from novel viruses. Through manual examination and validation of the 10 longest scaffolds, we successfully recovered 4 complete viral genomes, 2 are novel as they cannot be found in the existing databases and the other 2 are related to known phages. This hybrid reference-based and de novo assembly approach used by FVE-novel represents a powerful new approach for uncovering near-complete viral genomes in metagenomic data.
1. INTRODUCTION
Viruses/phages (or bacteriophages whose hosts are bacteria or archaea, hereafter used interchangeably) are the most abundant biological entities on Earth and our recognition of their ubiquitous presence and enormous diversity has been made possible only recently by culture-independent high-throughput sequencing (DeLong, 2009; Hanson et al, 2012; Hug, 2018; Pace, 1997; Rappé and Giovannoni, 2003; Roux et al, 2015; Whitman et al, 1998; Zou et al, 2016).
Viruses are now increasingly realized as a major player of the global ecosystem (Roux et al, 2016; Suttle, 2007) and a key biotic component influencing climate change (Danovaro et al, 2011; Puxty et al, 2016). However, despite their significance, our understanding is greatly limited by the vast unknown sequence space, >99% of the viruses are uncharacterized for their genome sequences and considered part of the “microbial dark matter” (Brum and Sullivan, 2015; Brum et al, 2016; Mizuno et al, 2013; Reyes et al, 2012; Roux et al, 2016; Villarreal and Witzany, 2010).
With the fast development of biotechnology, recovering unknown/novel viral genomes is now made possible by the increasing availability of metagenomic data. The most commonly used approach is to conduct de novo assembly of metagenomic reads followed by sequence comparison to determine whether or not the contigs assembled are novel. However, using de novo assembly alone is highly insufficient for recovering complete or near-complete unknown genomes [i.e., contigs that cover >90% of the expected genome sizes (Roux et al, 2019)]. For example, a large-scale marine metagenomic study showed that <0.2% of the set of 27,346 viral contigs generated by de novo assembly are complete (Coutinho et al, 2017).
Another study found that de novo assembly of metagenomic short reads generated from marine environmental samples could not fully recover any of the 1000 full-length phage genomes revealed by single-molecule nanopore sequencing of the same samples (Beaulaurier et al, 2020). Thus, de novo short read assembly methods alone are inadequate for recovering the complete genomes of unknown viruses/phages from metagenomic data.
To address the limitation, we develop FastViromeExplorer (FVE)-novel, a computational pipeline that combines short read mapping, de novo assembly, and iterative local assembly together to reconstruct complete or near-complete viral genomes. FVE-novel leverages FVE (Tithi et al, 2018), a program we developed previously that uses a pseudoalignment approach, to quickly align reads to a database of viral sequences to identify the presence and relative abundance of known viral groups in a metagenomic sample. FVE-novel calls FVE to identify viral reads from metagenomic data. It then uses SPAdes (Bankevich et al, 2012; Nurk et al, 2017a) to assemble the viral reads to generate viral contigs/scaffolds. The mapping step allows the removal of nonviral reads, making the assembly faster than the commonly used approaches that usually assemble all the reads in the sample.
Next FVE-novel grows each scaffold using iterative local assembly to generate final viral scaffolds. Those scaffolds are then compared with the input references, and the average nucleotide identity (ANI) is used to determine whether the viral scaffolds are potentially novel. FVE-novel reports potential novel viral scaffolds together with per base depth of coverage, which can be used to examine the quality of the generated scaffolds. FVE-novel is freely available at https://github.com/saima-tithi/FVE-novel
2. METHODS
2.1. Algorithm overview
The inputs of FVE-novel are metagenomic short reads and a reference database of genomic and/or contig sequences. The FVE-novel pipeline contains three main steps, (1) the read mapping and seed scaffold generation step, (2) the scaffold extension step where the seed scaffolds are extended through iterative local assembly, and (3) generating ANI and coverage statistics for all extended scaffolds. Figure 1 shows the workflow of all the steps.
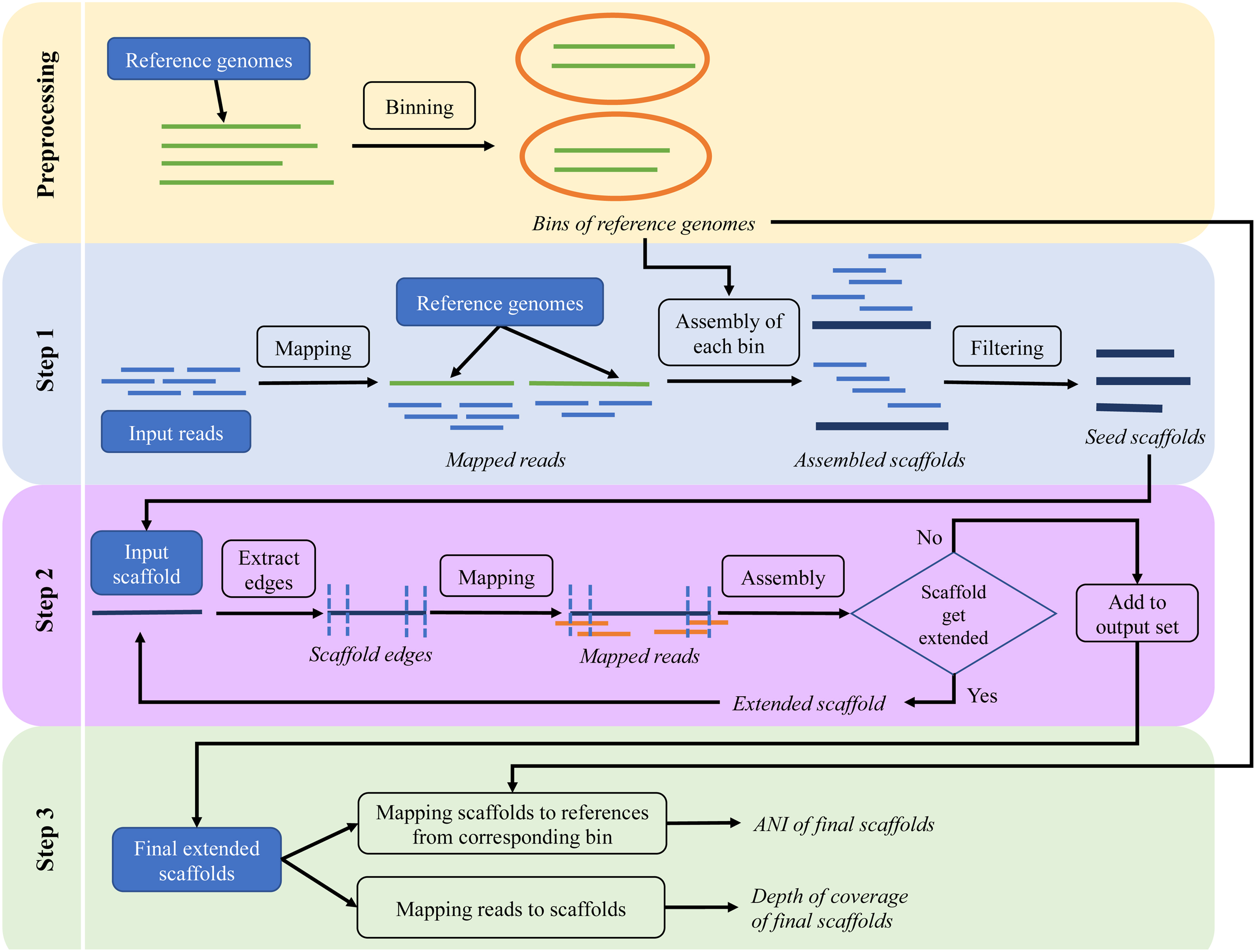
Overview of the FVE-novel pipeline, where the inputs are single-end or paired-end reads, and reference database and the output are a set of final extended scaffolds along with ANI and depth of coverage of the output scaffolds. ANI, average nucleotide identity; FVE, FastViromeExplorer.
2.2. Preprocessing the reference database
As reference databases often contain highly similar sequences, FVE-novel includes a preprocessing step that bins or clusters the reference sequences into groups using CD-HIT (Fu et al, 2012) with 95% similarity cutoff (users can change this cutoff score). This step is optional and may be unnecessary depending on the reference database users choose (e.g., the step can be bypassed if the selected database has already been binned).
2.2.1. Step 1. Read mapping and seed scaffold generation
In the first step, FVE-novel takes single- or paired-end read data and the reference database as input and invokes FVE (Tithi et al, 2018) to map the reads to the reference sequences. As metagenomic data contain reads from various organisms, using FVE, all the reads coming from bacteria, archaea, and other hosts are quickly filtered out and only viral reads are retained for the next assembly step. In this way, assembling genomes other than viruses can be avoided, making assembly much faster and more efficient.
Next, all the mapped reads by FVE are binned based on the binning of the reference genomes. In this study, all the reads mapped to the reference contigs/genomes from the same bin are pooled together. Based on the type of input reads, either SPAdes [(Bankevich et al, 2012), for single-end reads] or metaSPAdes [(Nurk et al, 2017b), for paired-end reads] can be used with default settings to assemble the reads into contigs/scaffolds. Both SPAdes and metaSPAdes break the reads into fixed-length k-mers, build de Bruijn graphs using the overlap of k-mers, and traverse the graph to produce longer sequences or genome fragments.
After the initial assembly step, all the generated sequences are examined and only the ones longer than 2 kb are kept and used as “seed scaffolds” or input scaffolds for the next extension step. The 2 kb cutoff is implemented so that FVE-novel does not attempt to extend every short segment that can be time-consuming, yet may contribute little to the overall scaffolds. Nonetheless, users can adjust this parameter depending on their research needs.
2.2.2. Step 2. Extending seed scaffolds using iterative assembly
In the extension step, the start and end edges (default length is read length *1.5 and can be changed by users) of each seed scaffold are extracted using BEDTools (Quinlan and Hall, 2010). Salmon (Patro et al, 2017) is used to map all the reads to the two edges. SPAdes is then used for assembly using the original scaffold as “—trusted-contigs” and the reads mapped to the edges of the scaffold as input reads. The mapping-and-assembly step is run iteratively for each seed scaffold until the scaffold stops growing. All the extended scaffolds are then clustered using CD-HIT (Fu et al, 2012) with 95% global ANI, and only the longest one in the cluster is kept as the final extended scaffolds.
2.2.3. Step 3. Analysis of new contigs and comparison with reference sequences
The final step involves generating summary statistics for the scaffolds including ANI, percentage of aligned nucleotides, and per base depth of coverage. As each scaffold is assembled from the reads mapped to a set of reference genomes belonging to a bin, the reference genomes in the bin are used for calculating ANIs of the corresponding scaffold using the MUMmer's “dnadiff” program (Kurtz et al, 2004). The output of FVE-novel contains ANIs and the percentage of aligned nucleotides between the extended scaffold and each of its reference genomes. All the reads are mapped back to each scaffold using Bowtie2 (Langmead and Salzberg, 2012) and depth of coverage is calculated using SAMtools (Li et al, 2009).
Summary statistics can be used to evaluate the quality and the novelty of the scaffolds. For example, a low ANI with all the reference genomes indicates that the scaffold might be novel or unknown. If the depth of coverage is fairly uniform along the scaffold, it is less likely to be chimeric.
2.3. Benchmarking on real data
To examine the performance of FVE-novel, we downloaded from the NCBI Sequence Read Archive 12 metagenomic samples (SRX2912986, SRX2912968, SRX2912972, SRX2912964, SRX2912992, SRX2912996, SRX2912975, SRX2912979, SRX2912983, SRX2912998, SRX2913002, and SRX2912985). These samples correspond to 12 time points at the same location/station in an ocean metagenomic study (Aylward et al, 2017) (see description in Supplementary Table S1). FVE-novel was applied to the sample SRX2912986 to recover viral scaffolds and the other 11 samples were used to evaluate the scaffolds generated.
For the reference database, we downloaded 24,411 viral contigs generated by a Global Ocean Virome (GOV) study (Roux et al, 2016). This study cross-assembled short reads of a total of 104 metagenomic samples taken from different depths of the oceans globally and generated the 24,411 viral contigs that were further binned into 15,280 groups, corresponding to epipelagic and mesopelagic viral populations and the remaining corresponding to bathypelagic viral populations.
3. RESULTS
3.1. Length distribution of the FVE-novel scaffolds
The ocean virome sample SRX2912986 (labeled as Station 70) contains 18,471,506 paired-end reads with read length 151 bps. Using the GOV database containing 24,411 contigs as reference and the reads from Station 70 as input, FVE-novel generated 268 scaffolds. Figure 2 shows the length distribution of the scaffolds, ranging from 2026 to 193,112 bps, with a median length of 4561 bps. There are 66 scaffolds longer than 10 kb.
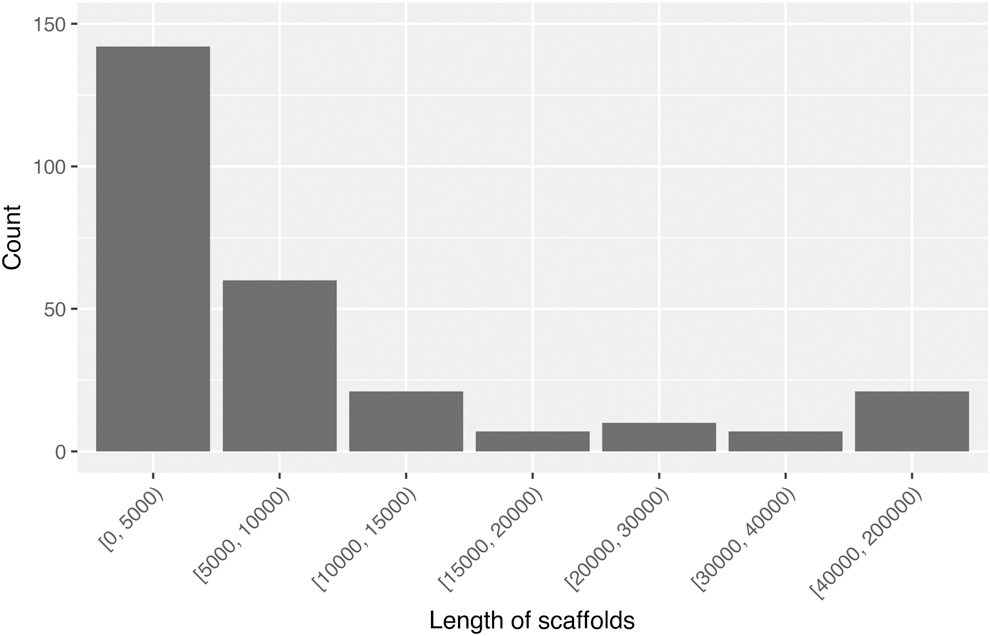
Length distribution of the 268 scaffolds generated by FVE-novel for the ocean metagenomic sample.
3.2. Comparison between the FVE-novel scaffolds and their reference sequences
As the scaffolds are originally assembled from the reads that are mapped to a set of contigs or genomes, these contigs or genomes can be considered the “reference contigs/genomes” of the scaffolds. Among the 268 scaffolds, 59 are longer than their corresponding reference sequences. Figure 3 shows the lengths of the 59 scaffolds with respect to those of their reference genomes, indicating that FVE-novel can generate considerably longer scaffolds than the original references. Moreover, some scaffolds have high ANIs (e.g., >95%) to their references, suggesting that parts of these scaffolds are present in the reference database and FVE-novel successfully extended those partial references. Some scaffolds have low ANIs to their references, indicating that these scaffolds are potentially novel.

The length comparison of the 59 scaffolds against their corresponding references (GOV database). These 59 scaffolds are a subset of the total 268 scaffolds, which are extended by the FVE-novel tool. The dotted line represents a 1:1 ratio between the x and y axis. The color of the dots represents the ANI between the scaffolds produced by FVE-novel and their references. GOV, Global Ocean Virome.
3.3. Comparison of the FVE-novel scaffolds against several databases
To examine the quality of the scaffolds generated by FVE-novel, we analyzed the longest 10 scaffolds in detail. The 10 scaffolds range from 72,532 to 193,112 bps, with average depths of coverage ranging from 48.5 × to 338 × .
As the scaffolds are assembled using the GOV database as the input reference database, it is expected that the scaffolds have sequence similarity to the GOV database sequences. Table 1 shows the BLAST results against the GOV sequences, with average ANIs (%) ranging from 89.21 to 97.78 and alignment percentage from 6.25 to 99.98. Figure 4, generated using Artemis (Carver et al, 2011), shows how the scaffolds align to their corresponding references in the GOV database and the sequence similarity, further supporting that FVE-novel successfully extended the reference genomes.

Alignment of the 10 scaffolds (
The Longest 10 Scaffolds of the 268 Scaffolds Generated by Applying FastViromeExplorer-Novel to the Ocean Metagenomic Sample Collected from Aylward et al (2017) Showing Average Nucleotide Identity and Aligned Nucleotide Percentage Between the Reference (GOV Database) Scaffold and the Extended Scaffold
These 10 scaffolds are also compared with the 483 scaffolds assembled in the original Aylward et al (2017) study and blast nr nucleotide database using Blast, the best hit from Blast is selected.
ANI, average nucleotide identity; GOV, Global Ocean Virome.
To see how the FVE-novel scaffolds compared with the 483 scaffolds reported previously in the original study (Aylward et al, 2017) from where we collected our read sample, we used BLASTN (Altschul et al, 1997) to compare FVE-novel generated scaffolds with the 483 assembled scaffolds. Table 1 shows the best hit result. Nine FVE-novel scaffolds have very high ANIs to their best hits (95.25% to 99.73%). This is not surprising as the same read sample was used to generate both the FVE-novel scaffolds and also some of the 483 scaffolds.
Interestingly, scaffold S6 does not have any hits in the 483 scaffolds, suggesting that FVE-novel was able to reconstruct a viral scaffold that was not recovered by the original study (Aylward et al, 2017). Scaffold 6 has a best hit to the GOV sequence with 97.78% ANI and 93.09% alignment length. Notably, all nine scaffolds generated by FVE-novel are longer than the scaffolds from the original study.
We also used BLASTN to compare the 10 scaffolds against the nr nucleotide database and found that 4 scaffolds (S0, S1, S2, and S6) have no significant hits and the other 6 scaffolds (S3, S4, S5, S7, S8, and S9) have similarity to the Prochlorococcus phage P-SSM4 (ANIs ranging from 86.46% to 92.68%) and the Prochlorococcus phage P-HM2 (ANI 84.89%). This result is consistent with the original study (Aylward et al, 2017) where parts of the Prochlorococcus phage genomes were also recovered from the data.
3.4. Comparison within the FVE-novel scaffolds
The observation that some of the 10 scaffolds have similar BLAST hits suggests that their sequences might be similar. Therefore, we also compared the scaffolds against each other (Fig. 5). Four groups of scaffolds are observed: group 1 containing S0, S1, and S6, group 2 containing only S2 with no significant similarity to any other nine scaffolds, group 3 containing S3, S4, S5, S7, and S9, and group 4 containing only S8. As FVE-novel removed all scaffolds except one with an identity greater than 95%, these scaffolds have similarity <95% and could be different viral species or divergent strains of the same virus. Note that 95% identity has been commonly used in the literature as the cutoff score for clustering/binning viral contigs into viral operational taxonomic units (vOTUs) (Gregory et al, 2019; Paez-Espino et al, 2016; Roux et al, 2016).

Percentage of similarity between each pair of the longest ten scaffolds of the 268 scaffolds generated by applying FVE-novel to the ocean metagenomic sample.
Group 3 scaffolds (S3, S4, S5, S7, and S9) all have similarity to the Prochlorococcus phage P-SSM4 (Table 1), thus likely represent different strains of Prochlorococcus phage P-SSM4. In the following, we analyzed the four groups of scaffolds in detail.
3.4.1. Group 1 scaffolds (S0, S1, S6)
S0 is the longest scaffold in group 1 and has a length 193,112 bps. It is generated from the Station 70 sample, and thus, we checked if this scaffold is also present in the other 11 viral metagenomic samples. Figure 6a shows the depth of coverage of S0 in all 12 samples. The average depth of coverage of S0 in Station 6, 14, 18, 22, 28, 32, 37, 52, 56, 61, 67, and 70 samples are 95.26, 28.39, 32.24, 94.78, 52.08, 73.75, 17.71, 20.18, 82.55, 148.61, 85.54, and 93.02, respectively, with Station 37 having the lowest coverage (17.71) and Station 61 (148.61) the highest coverage. The pairwise Pearson correlation coefficient of per base coverage of S0 between any two samples ranges from 0.77 to 0.94, and therefore, even though the abundances of S0 differ among stations, the per base read coverages along the scaffold are highly correlated among samples.
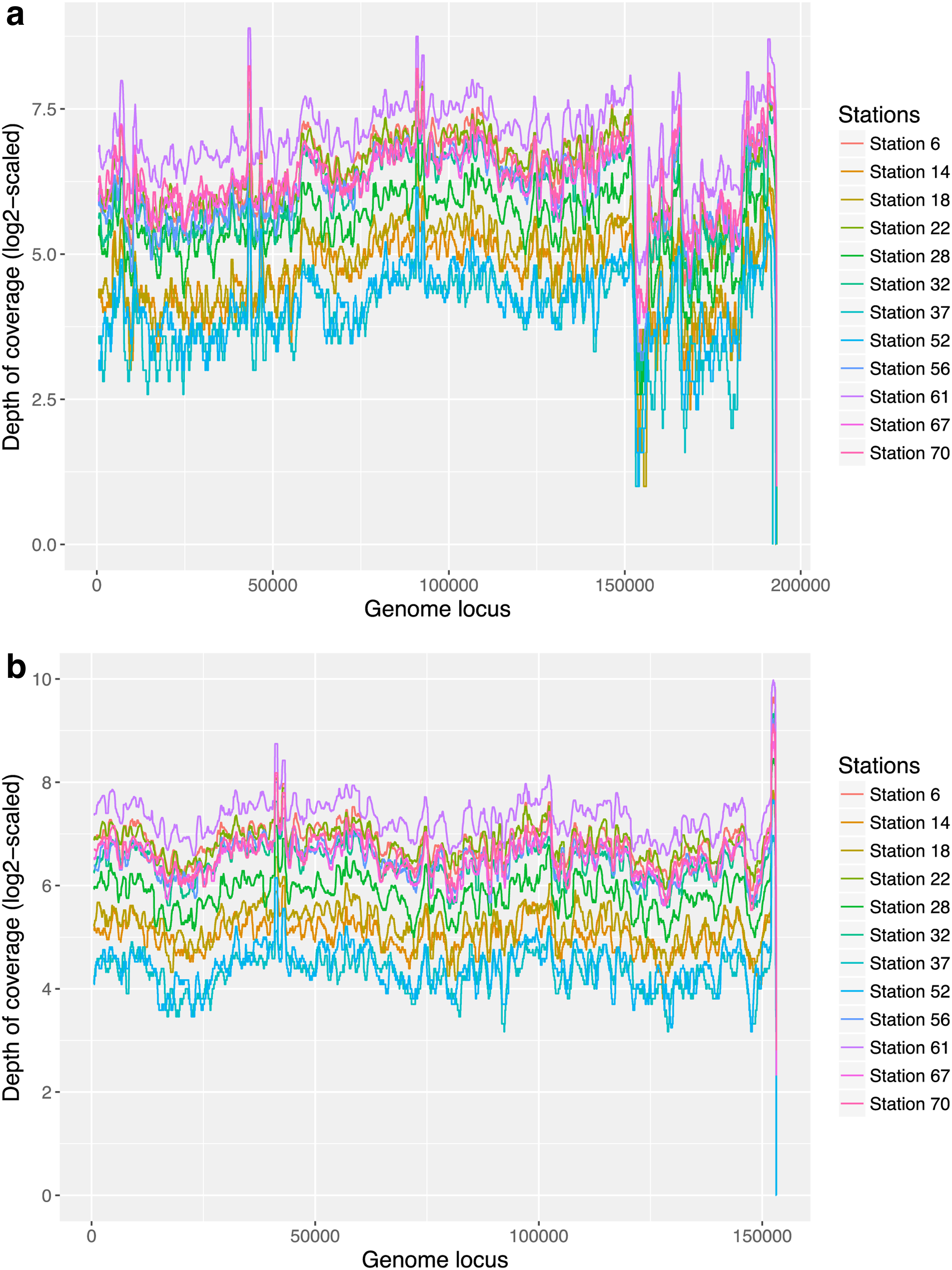
The log2-scaled depth of coverage of
Figure 6a also shows that coverage dropped greatly after 150 kb for all samples, which means fewer reads got mapped to this region. We further analyzed this region for sequence composition and did not find any homopolymer or short repeats, and thus, the lower mapping rate is not caused by repeat sequences. Another possible cause of coverage drop is that this part of the scaffold was the misassembly from a less abundant strain of the same virus. In this study, we explored this possibility by reassembling the scaffold. Specifically, we used the “Map to Reference” algorithm implemented in Geneious 11.0.4 (Kearse et al, 2012) to examine whether there are multiple viral strains and if there are, whether a complete assembly of the dominant strain can be generated. All of the metagenomic reads in the Station 70 sample were aligned to scaffold S0 using the “Low sensitivity/Fastest” settings allowing for 10% mismatches. The alignment showed a large number of single-nucleotide polymorphisms, revealing the existence of two or more strains for this phage as well as distinct regions of high and low coverages consistent with the suspected chimeric assembly of these strains. To recover the dominant strain, the consensus sequence from the alignment was segmented into contigs with high coverage >40 × . These contigs were binned into lists of contigs with similar coverage for further assembly. Next, contigs in each bin were iteratively extended using Geneious by mapping reads to the contig ends with high stringency. Specifically, all of the paired-end reads that were previously mapped to the contigs were aligned to these high-coverage contigs using “Map to Reference” with stringent “Custom Sensitivity” settings allowing no >1% “Mismatches per Read” and 1% “Gaps per Read” and requiring that both of the paired-end reads map to the new consensus sequence. This process was iterated for each contig using Geneious' “Fine Tuning” settings up to “100 times.” This process was continued until the extended contigs merged together, maintaining approximately uniform coverage, and could no longer be extended or closed into a circular genome sequence. Finally, we recovered a 153 kb scaffold from scaffold S0 with uniform coverage across all 12 samples (Fig. 6b).
Comparison of this 153 kb scaffold with S0 reveals that the middle 90 kb of S0 (starting at 60 kb and ending at 150 kb) is exactly the same as the 153 kb strain assembled by Geneious (Supplementary Fig. S1). However, the first 60 kb of S0 has around 80% similarity to the 153 kb strain, and so, this part of S0 could be from a different strain and the last 40 kb of S0 (staring at 150 kb and ending at 193 kb) is the result of assembly artifact (Supplementary Fig. S1). Comparison of the 153 kb strain with S1 (155 kb) shows that FVE-novel recovered this dominant strain successfully in S1 (Supplementary Fig. S2).
As scaffold S1 is a correct representation of the dominant strain of this novel virus, from Figure 7, we can see that S1 has a uniform coverage across all 12 samples. In scaffold S6, our tool captured the 80 kbp of the 153 kbp dominant strain (Supplementary Fig. S3) and S6 also has a uniform depth of coverage across all 12 samples (Supplementary Fig. S4). To find out if this virus has multiple strains present in the Station 70 sample, we subsampled the 153 kb strain into 10 pieces of 20 kb long and applied TenSQR (Ahn et al, 2018), a viral quasispecies reconstruction tool, to each piece. For each piece, TenSQR reported 2 or 3 strains, consistent with our observation that multiple strains of this virus are present in the Station 70 sample (Supplementary Table S2).
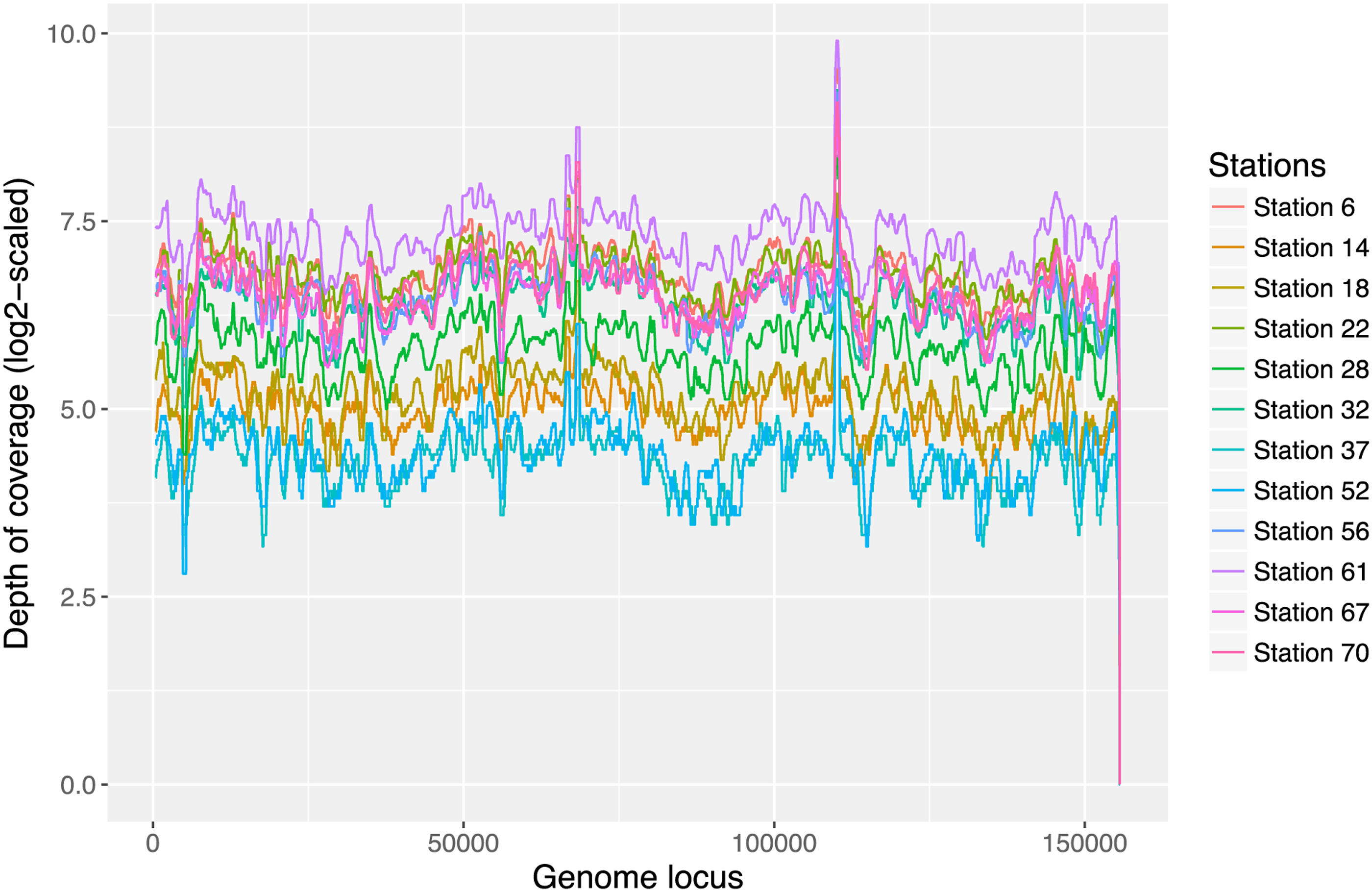
The log2-scaled depth of coverage of S1 (155 kb) across 12 ocean samples (showing median coverage of per 1000 bp window with step size 1).
3.4.2. Group 2 scaffold (S2)
Scaffold S2 is present in all samples with varying abundances but highly correlated depth of coverage among the samples (Fig. 8a). With the same aforementioned procedure, we generated a 151 kb scaffold that most likely represents a complete novel virus genome recovered from S2. Figure 8b shows that this 151 kb scaffold has a uniform depth of coverage across all 12 samples. Comparison of this scaffold with S2 shows that our tool successfully recovered most of this virus except a 15 kb long portion of it (Supplementary Fig. S5). We also checked if multiple strains of this virus are present in the Station 70 sample using TenSQR. Results show that multiple strains of this virus are also present in this sample (Supplementary Table S3).
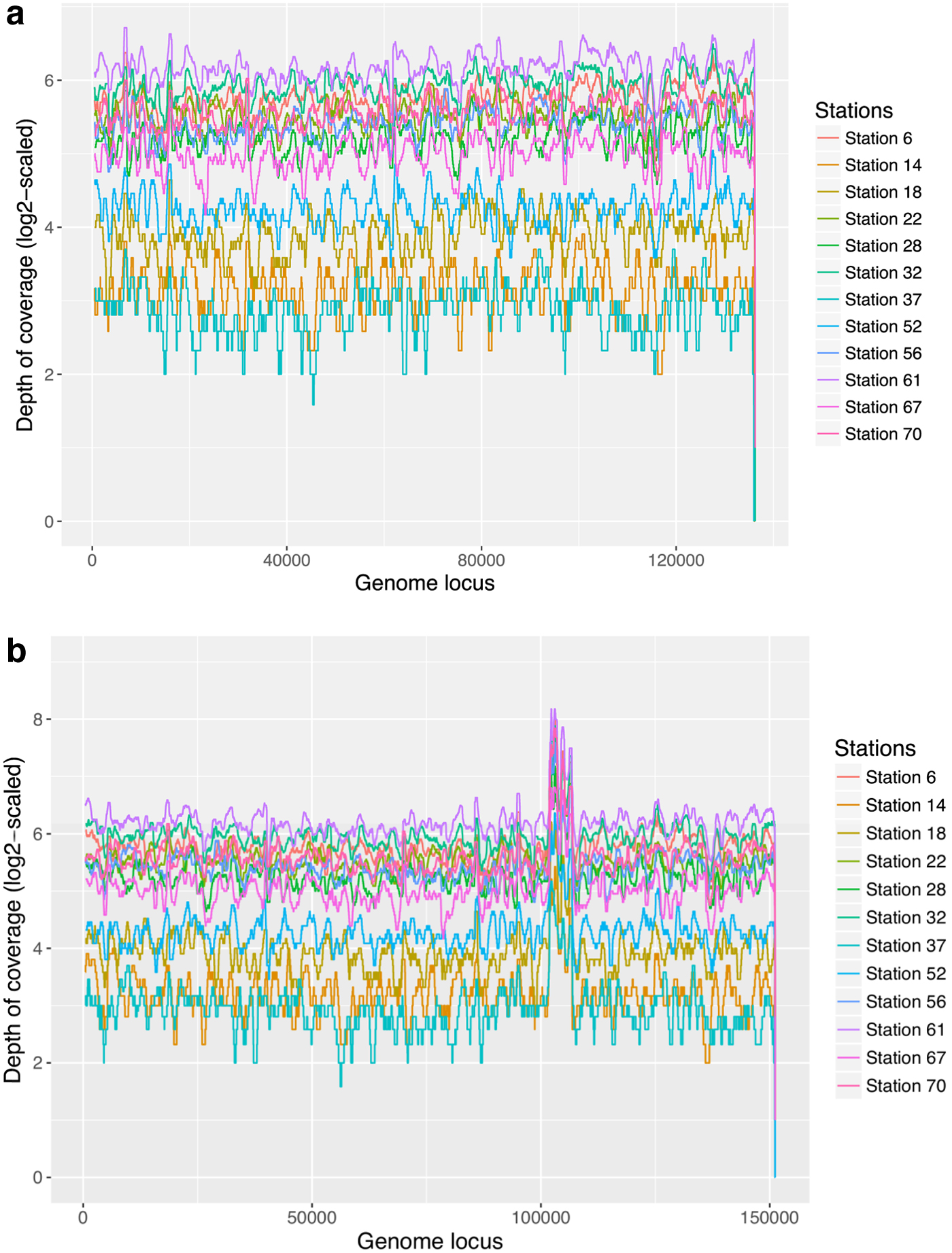
The log2-scaled depth of coverage of
3.4.3. Group 3 scaffolds (S3, S4, S5, S7, S9)
As scaffolds S3, S4, S5, S7, and S9 have similarity to the Prochlorococcus phage P-SSM4, we chose to analyze in detail the longest scaffold S3 with length 132,604 bp. Figure 9a shows consistent depth of coverage of S3 across all 12 samples, but coverage dropped dramatically in all samples from 50 to 65 kb. We then aligned all the reads from Station 70 sample to scaffold S3 using Geneious and observed the presence of multiple strains. Using the same procedure as above, we were able to recover a 177 kb scaffold representing the dominant strain of Prochlorococcus phage P-SSM4 present in Station 70 sample, with a uniform coverage across all 12 samples (Fig. 9b).

The log2-scaled depth of coverage of
Comparison of this 177 kb scaffold with S3 shows that S3 matches the dominant strain with about 100% similarity for all except the 15 kb segment (the 50–65 kb part in S3) where the similarity dropped to 80% (Supplementary Fig. S6). This result implies that our tool successfully captured about 117 kb of the dominant strain except from a piece of length 15 kb where the assembly switched to a different strain with lower coverage. Consistently, analysis with TenSQR suggests the presence of three to seven strains in the sample (Supplementary Table S4). Interestingly, alignment of the 177 kp scaffold to Prochlorococcus phage P-SSM4 (length 178,249 bp) shows similar as well as more divergent regions (ANI 82.9%) (Supplementary Fig. S7).
3.4.4. Group 4 scaffold (S8)
The depth of coverage of the scaffold S8 is quite uniform across all 12 samples along the region except the beginning 6 kb and the last 10 kb where coverage is higher than the remaining region, which can be caused by a different strain (Fig. 10a). According to the blast result, S8 has similarity to the Prochlorococcus phage P-HM2. We then used Geneious to recover the dominant strain from S8 and obtained a 183 kb scaffold. Figure 10b shows that the 183 kb scaffold has uniform coverage across all 12 samples. Comparison with S8 shows that in S8 we recovered about 60 kb of this dominant strain (Supplementary Fig. S8). Analysis with TenSQR suggests the presence of two or three strains in this sample. Alignment of the 183 kb scaffold to Prochlorococcus phage P-HM2 (length 183,806 bp) also shows many similar regions and some dissimilar regions (ANI 86.56%) (Supplementary Fig. S9).
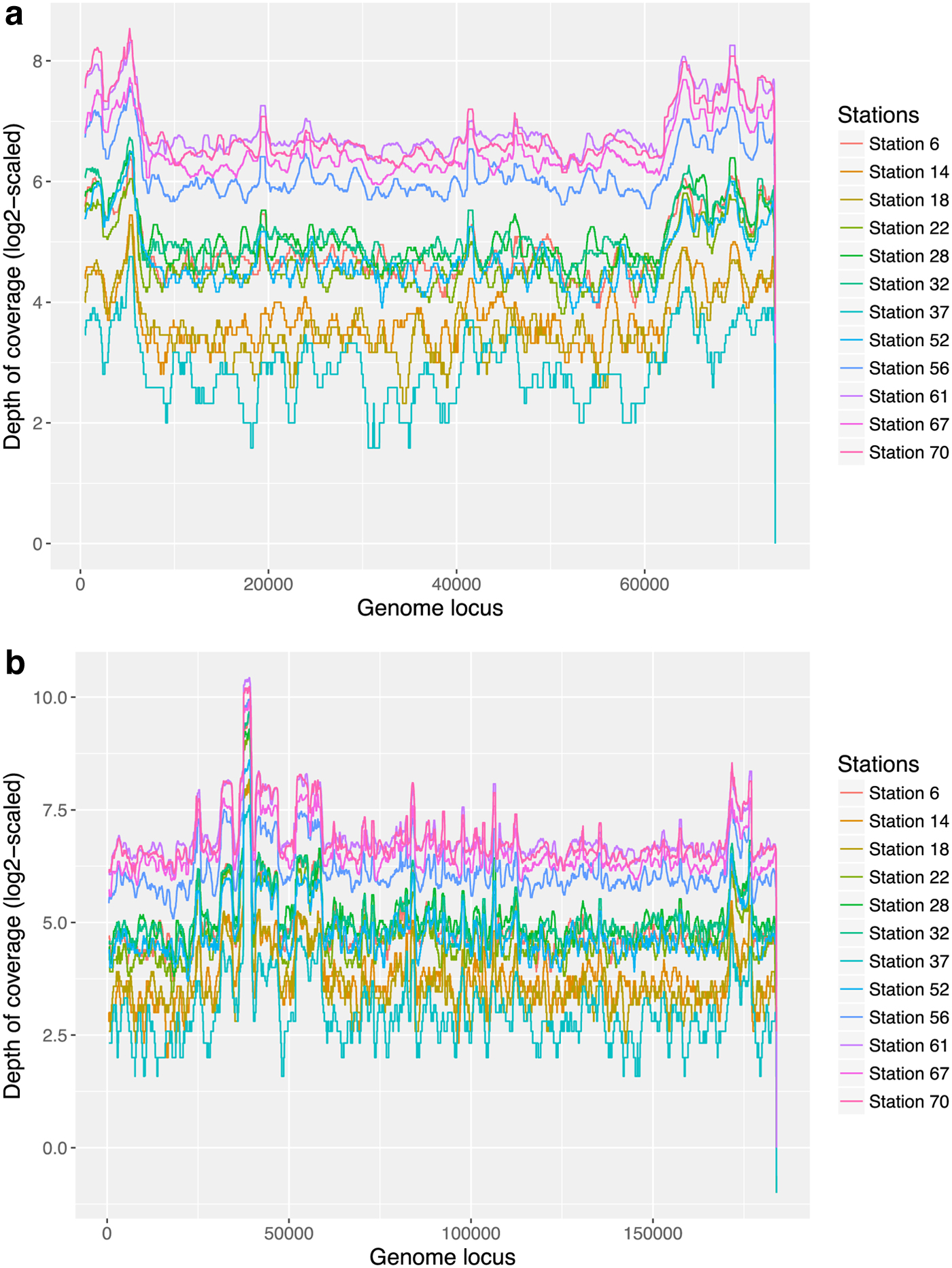
The log2-scaled depth of coverage of
3.5. Function annotation of the four complete scaffolds
We annotated genes from the four completed scaffolds: (1) the 153 kb dominant strain of a novel virus recovered from S0, (2) the 151 kb dominant strain of a novel virus recovered from S2, (3) the 177 kb dominant strain of Prochlorococcus phage P-SSM4 recovered from S3, and (4) the 183 kb dominant strain of Prochlorococcus phage P-HM2 recovered from S8. The four scaffolds have 195, 160, 220, and 231 proteins, respectively, predicted by Prodigal with metagenomic mode (i.e., Prodigal option: -p meta) (Hyatt et al, 2010). We then annotated those proteins using eggNOG-mapper with the virus database and HMMER option (Huerta-Cepas et al, 2017).
For all the four scaffolds, eggNOG-mapper annotated some viral structural proteins such as capsid, baseplate, and virion, indicating that they are potential virus genomes. Figure 11 shows the protein annotation of the two novel genomes recovered from S0 and S2. Figure 12 shows the protein annotation of the two Prochlorococcus phage strains recovered from S3 and S8, respectively. As both phages are Prochlorococcus phage strains, as expected, the arrangement of viral structural proteins in these phage genomes is similar to that of Prochlorococcus.

Protein annotation of the two novel scaffolds:
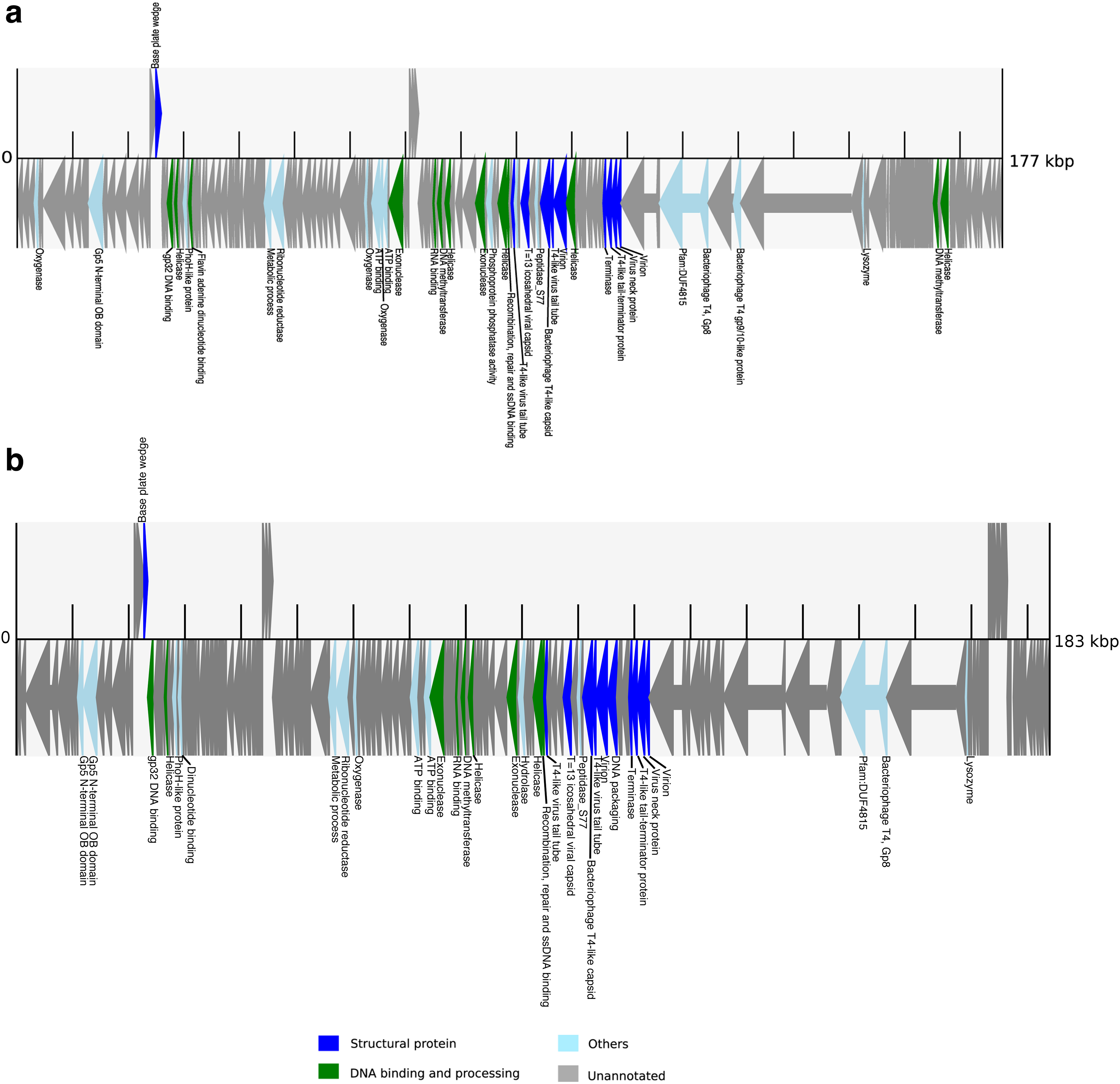
Protein annotation of the two strains of Prochlorococcus phage:
4. DISCUSSION
In this article, we present FVE-novel, a computational pipeline for recovering viral scaffolds based on reference-based mapping and iterative assembly. By applying our tool to an ocean metagenomic sample, we assembled 268 viral scaffolds. Some of these viral scaffolds are quite long, which can be potential near-complete viral genomes. Manual curation and validation of the 10 longest scaffolds led to successful recovery of 4 complete viral genomes. Among these four viral genomes, two are novel genomes as they were not found in the existing databases, one represents strain of Prochlorococcus phage P-SSM4 and another one represents strain of Prochlorococcus phage P-HM2. We also noted substantial microdiversity in the phage genomes present in the metagenomic data, which should be considered further in future work, given that it is a potential complication to the recovery of full-length viral genomes.
Out of the 18,471,506 paired-end reads from Station 70 sample, 1,240,164 reads (∼6.7%) were mapped to the 268 scaffolds with 8.6% mapped to the 153 kb novel virus recovered from S0, 4.2% mapped to the 151 kb novel virus recovered from S2, 35.3% mapped to the 177 kb dominant strain of Prochlorococcus phage P-SSM4, and 13.9% mapped to the 183 kb strain of Prochlorococcus phage P-HM2. Thus, the four new viruses contribute to >62% of the mapped reads, indicating that they are very abundant in Station 70 sample.
Assembly of abundant marine viruses revealed a high incidence of microdiversity in naturally occurring viral populations in the environment. Genomic chimeras often have anomalous coverage since they represent distinct genotypes that have been incorrectly merged, and most metagenomic assemblers use the coverage information to break contigs or scaffolds into smaller pieces to avoid misassembly (García-López et al, 2015; Namiki et al, 2012; Nurk et al, 2017a; Vázquez-Castellanos et al, 2014). Given that the recovery of complete viral genomes from metagenomic assemblies is rare, it is plausible that microdiversity may be a prevalent cause of assembly fragmentation. Moreover, given that genomic microdiversity is difficult to identify and may not always lead to contig breakage by assembler, it is possible that a substantial number of viral contigs or scaffolds present in publicly available repositories are in fact chimeras of multiple viral strains.
The prevalence of this is difficult to ascertain given the paucity of high-quality complete viral genomic references for benchmarking. Future studies should therefore prioritize the rigorous identification of strain-level microdiversity in viral populations to identify the nature and extent of this genome-wide microdiversity in nature.
5. CONCLUSIONS AND USAGE NOTES
Overall, FVE-novel implements a novel strategy to recover viral genomes and will serve as a powerful tool for future studies that will continue to enhance existing viral databases. The three steps in the FVE-novel pipeline (Fig. 1) are implemented as separate modules so that users can use each module separately if needed. For example, for any metagenomic data, users can run the entire pipeline, that is, all three steps, to obtain viral scaffolds. Alternatively, if a user has a read sample and some viral sequences generated from those reads (e.g., using a different assembler), the user can directly run the second seed extension step to extend the input viral sequences (all the modules are freely available at https://github.com/saima-tithi/FVE-novel).
For each scaffold generated, FVE-novel reports the ANI and percentage of aligned nucleotide between the scaffold and its reference. FVE-novel also reports per base depth of coverage along the scaffold, which can be used similarly to what we have shown above for examining the scaffold quality and identifying “suspicious regions” of misassembly. As our detailed analyses show that having multiple strains of the same viruses can create great challenges to assembly programs, it is paramount that users examine the report on depth of coverage for the generated scaffolds to ensure the quality of the putative novel viral genomic sequences.
Footnotes
AUTHORs' CONTRIBUTIONS
S.S.T. and L.Z. conceived and designed the experiments. S.S.T. performed the experiments, implemented the software, wrote the article, and prepared the figures and tables. All authors contributed to the analysis of data and review of the article before submission for publication. All authors read and approved the final article.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
FVE-novel is freely available to download at https://github.com/saima-tithi/FVE-novel. The 12 ocean microbiome samples used in our study are publicly available in the NCBI SRA under the accession numbers SRX2912986, SRX2912968, SRX2912972, SRX2912964, SRX2912992, SRX2912996, SRX2912975, SRX2912979, SRX2912983, SRX2912998, SRX2913002, and SRX2912985 and these samples were originally prepared and analyzed by Aylward et al (![]() ).
).
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
Publication of this article was partially funded by the Virginia Tech's Open Access Subvention Fund.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.