
Abstract
High-throughput DNA and RNA sequencing are revolutionizing precision oncology, enabling personalized therapies such as cancer vaccines designed to target tumor-specific neoepitopes generated by somatic mutations expressed in cancer cells. Identification of these neoepitopes from next-generation sequencing data of clinical samples remains challenging and requires the use of complex bioinformatics pipelines. In this paper, we present GeNeo, a bioinformatics toolbox for genomics-guided neoepitope prediction. GeNeo includes a comprehensive set of tools for somatic variant calling and filtering, variant validation, and neoepitope prediction and filtering. For ease of use, GeNeo tools can be accessed via web-based interfaces deployed on a Galaxy portal publicly accessible at https://neo.engr.uconn.edu/. A virtual machine image for running GeNeo locally is also available to academic users upon request.
1. INTRODUCTION
Cancer-specific epitopes (referred to as neoepitopes), generated by nonsynonymous somatic single nucleotide variants (SNVs) carried by tumor cells, have become the main focus of personalized cancer immunotherapies. However, the identification of neoepitopes from next-generation sequencing (NGS) data in a clinical setting remains challenging and requires the use of complex bioinformatics pipelines. These pipelines must address a number of challenging computational problems, including accurate identification of somatic SNVs from short-read NGS data and predicting which mutated peptides get presented on the surface of tumor cells and can mediate antitumor immune responses.
Many of the existing bioinformatics tools focus on the epitope prediction step for a given set of somatic variants. Examples of such tools are CloudNeo (Bais et al, 2017), NeoEpitopePred (Chang et al, 2017), Neoepiscope (Wood et al, 2020), and Mupexie (Bjerregaard et al, 2017). Neopepsee (Kim et al, 2018) and NeoPredPipe (Schenck et al, 2019) also take somatic variants as input and predict T-cell receptor (TCR) recognition potential for the corresponding mutated peptides. There are only a few neoepitope prediction pipelines that integrate variant calling from matched tumor/normal NGS data, including the tumour-specific neoantigen detector (TSNAD) (Zhou et al, 2017), pTuneos (Zhou et al, 2019), DeepAntigen (Shi et al, 2020), ProTECT (Rao et al, 2020), Epidisco (Rubinsteyn et al, 2018), and OpenVax (Kodysh and Rubinsteyn, 2020).
In this paper, we present a new suite of tools, referred to as GeNeo, for predicting neoepitopes from matched tumor/normal exome sequencing data coupled with tumor RNA-Seq data. A distinguishing feature of GeNeo is that it can analyze multi-technology sequencing data generated using the Illumina and Ion Torrent platforms to take advantage of the complementary strengths of the two technologies (Boland et al, 2013). Combined with a consensus variant calling approach that integrates two different variant callers, Strelka (Saunders et al, 2012) and SNVQ (Duan et al, 2014), this results in highly accurate somatic variant calls. To facilitate use within a clinical setting, where best practices require validation of somatic SNVs by an orthogonal technology (Koboldt, 2020), GeNeo includes tools enabling high-throughput validation by targeted resequencing using the AccessArray platform. Finally, GeNeo includes tools for predicting neoepitopes encoded by validated somatic SNVs and prioritizing them using a variety of criteria including their major histocompatibility complex class I (MHC-I) binding affinity and differential agretopic index (DAI), which was previously shown to be a better predictor of tumor control in vaccination experiments in mouse tumor models (Duan et al, 2014).
The GeNeo tools have been used in several clinical studies, including phase I and II clinical trials. The tools have been deployed using a customized instance of the Galaxy framework (Afgan et al, 2018) and are publicly accessible through easy-to-use graphical user interfaces at https://neo.engr.uconn.edu/. A virtual machine image for running GeNeo locally is also available to academic users upon request.
2. METHODS
In this section, we describe the tools included in the GeNeo toolbox (Fig. 1). The tools are grouped into three categories: variant calling and filtering, variant validation, and neoepitope prediction.
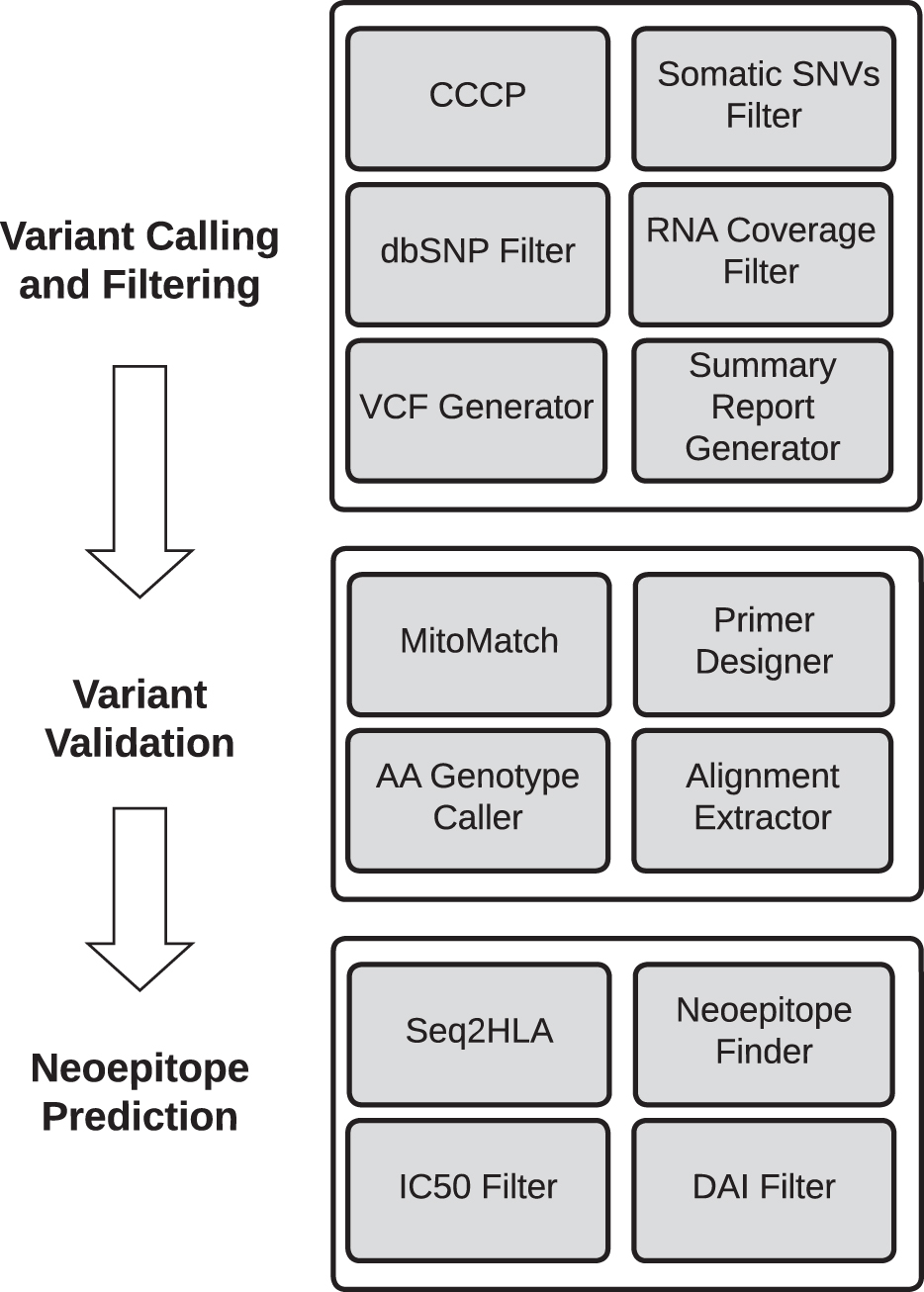
The tools comprising the GeNeo toolbox are grouped into three categories. The variant calling and filtering category includes the CCCP tool along with tools for filtering SNVs based on different criteria, generating a summary report, and converting the (filtered) CCCP output to the standard VCF for analysis by external tools. The variant validation category includes tools for identifying possible sample mismatches based on mitochondrial haplogroups, designing primers for targeted resequencing of candidate SNVs using the AccessArray platform, demultiplexing AccessArray amplicon sequencing data and calling SNV genotypes, and extracting read alignments that cover candidate SNVs for manual review. The neoepitope prediction category includes tools for inferring HLA alleles from RNA-Seq data, predicting neoepitopes, and filtering them based on predicted IC50 and DAI values. AA, AccessArray; CCCP, Consensus Caller Cross-Platform; DAI, Differential Agretopic Index; dbSNP, the Single Nucleotide Polymorphism database; HLA; IC50, half maximal inhibitory concentration; SNVs, single nucleotide variants; VCF, variant calling format.
2.1. Variant calling and filtering tools
Variant calling is a critical first step of any immunogenomic analysis. Because the focus of these analyses is on the identification of neoepitopes encoded by somatic mutations present in the patient's tumor, variant calling is typically performed using deep whole-exome sequencing data generated from matched tumor and normal samples. Processing the large volume of sequencing data requires efficient algorithms and computing infrastructure able to deliver results in a timely manner. At the same time, clinical samples can undergo unpredictable degrees of degradation, resulting in a high patient-to-patient variation in the quality and quantity of sequencing data. This requires complex, multistep analysis pipelines that are always able to produce reliable variant calls. Filtering somatic variant calls is also essential for eliminating from consideration subclonal mutations present in a minority of tumor cells that may generate poor immunotherapy targets as well as reducing the number of false positives generated by library preparation artifacts and sequencing errors. GeNeo implements the recommended best practices for somatic variant calling and filtering (Koboldt, 2020) as described hereunder.
2.1.1. Consensus caller cross-platform
There are multiple NGS technologies commercially available, each with its own strengths and weaknesses (Goodwin et al, 2016). Although single-molecule sequencing technologies have rapidly advanced over the past few years, their higher error rate makes them less suited for variant calling. Indeed, clinical sequencing is almost universally performed using short-read NGS technologies. These technologies deliver at a very low cost and with a lower error rate of millions of reads of a few hundred bases, sufficiently long to be reliably mapped against the genome. Among commercially available short-read NGS technologies, the Illumina and Ion Torrent platforms have comparable costs. Both technologies perform sequencing by synthesis. Illumina uses fluorophore-labeled single nucleotides with the 3′-OH group blocked in the synthesis step.
Their cyclic reversible termination process ensures that at most one nucleotide is incorporated in the synthesized DNA molecules in each cycle, with incorporation events being detected by high-resolution microscopy. This results in a low rate of homopolymer sequencing errors at the cost of increased sequencing time of several days for their high-end instruments. In contrast, Ion Torrent uses natural nucleotides in each synthesis cycle (with no need for extra steps for removing the 3′ blocking groups) and detects nucleotide incorporation using field-effect transistors sensitive to pH changes. This dramatically reduces sequencing time to a few hours but also results in limited sequencing accuracy of homopolymer repeats.
The Consensus Caller Cross-Platform (CCCP) tool was designed to leverage the complementary strengths of the Illumina and Ion Torrent technologies. As shown in Figure 2 by default the tool takes as input Illumina paired-end and Ion Torrent (single-end) reads generated from matched tumor and normal exomes, along with Illumina paired-end and Ion Torrent RNA-Seq reads, all in fastq format. However, the tool also has the option of analyzing Illumina single-end reads along with Ion Torrent reads, as well as single technology data (Illumina paired-end only, Illumina single-end only, or Ion Torrent only). In addition, although primarily designed for human sequencing data, CCCP has the option of analyzing tumor model data from the commonly used C57BL/6 and BALB/c mouse strains. The tool generates an unfiltered list of candidate SNVs along with extensive information on the allele coverage in the Illumina and Ion Torrent exome and RNA-Seq datasets and calls made by two somatic SNV callers, Strelka (Saunders et al, 2012) and SNVQ (Duitama et al, 2012), with subtraction. The output of CCCP is used as input to the provided filtering tools to identify high-confidence somatic calls, which can then be validated by targeted resequencing and used to predict neoepitopes.

The Galaxy interface of the CCCP tool. By default, the tool takes as input Illumina paired-end and Ion Torrent (single-end) reads generated from matched tumor and normal exomes, along with Illumina paired-end and Ion Torrent RNA-Seq reads.
Figure 3 provides the main steps implemented by the CCCP tool. Following recommended best practices for somatic variant calling (Koboldt, 2020), CCCP uses FastUniq (Xu et al, 2012) to remove duplicate reads, which are likely to be artifacts of polymerase chain reaction (PCR) amplification. This is particularly important for Ion Torrent exome sequencing data, which uses an exome capture protocol based on highly multiplexed PCR, but PCR artifacts can be generated during Illumina library preparation too. Removing duplicated reads reduces the likelihood that a PCR error that gets amplified and sequenced multiple times results in a false-positive call. Following duplicate removal, Trimmomatic (Bolger et al, 2014) is used to remove reads with an average quality score of <20 and reads shorter than a user-specified length (default 50). Subsequently, all exome and RNA-Seq reads passing the FastUniq and Trimmomatic filters are mapped against the genome using hierarchical indexing for spliced alignment of transcripts version 2 (HISAT2) (Kim et al, 2015), with spliced alignments disabled for exome reads. Alignments of nonuniquely mapped reads and nonconcordant pairs are discarded.
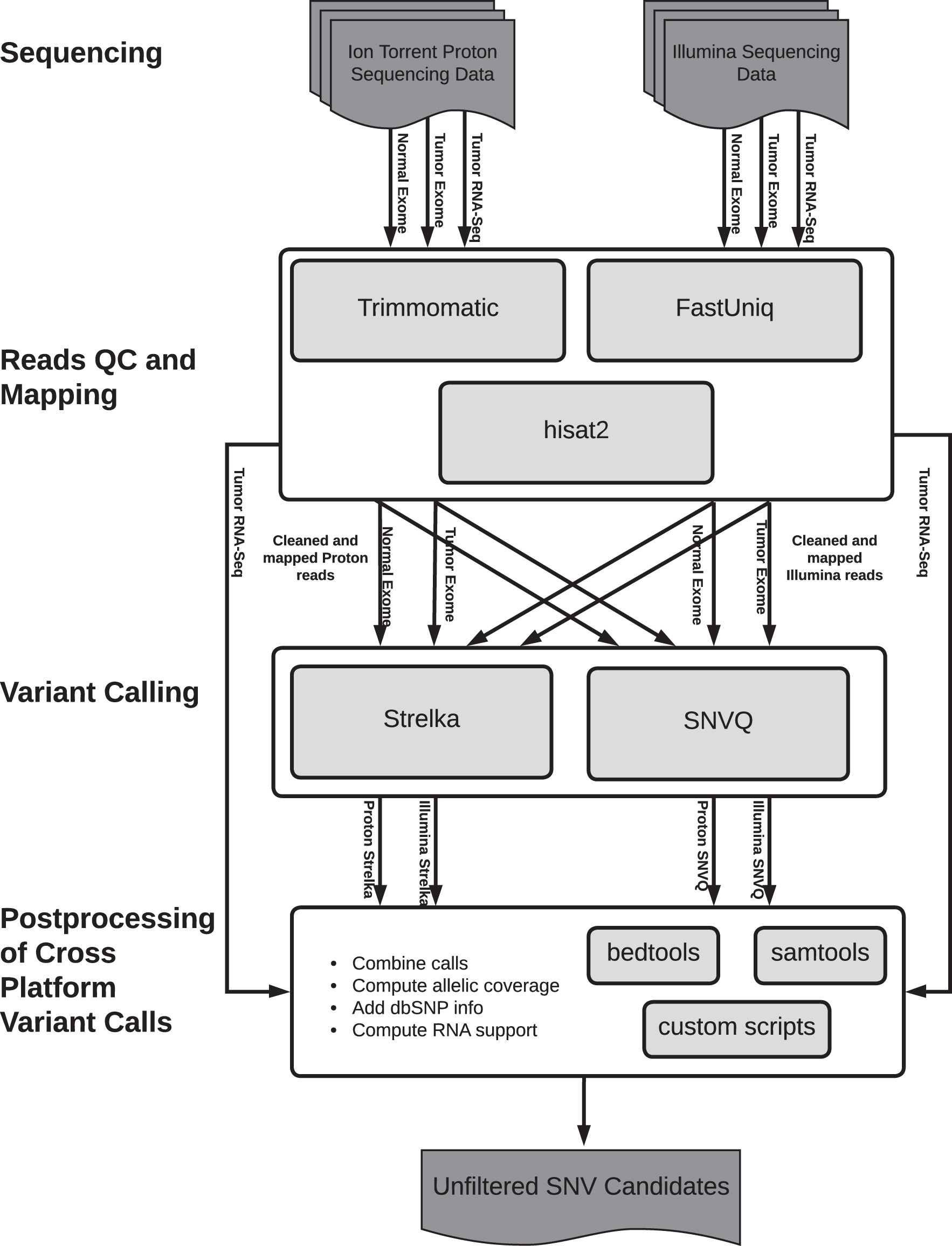
A flowchart of the CCCP tool for calling somatic variants. The CCCP output includes an unfiltered aggregation of SNV calls from both platforms and both callers. SNVQ.
Candidate somatic SNVs are then called by running two somatic SNV callers [Strelka, Saunders et al (2012) and SNVQ, Duitama et al (2012) with subtraction] on the alignments generated from each of the two sequencing technologies. Strelka is independently run on the alignments generated from Illumina and Ion Torrent-matched exomes using the “no depth filter” option. The SNVQ subtraction method is also used to process independently the alignments generated from Illumina and Ion Torrent exomes. For each technology, SNVQ is run separately to call the SNVs from the normal and tumor exome alignments. A high-confidence SNV call is defined as a call made with an SNVQ genotype Phred score of at least 50, alternative allele support coverage of at least 3 reads, and alternative allele support on both strands.
An SNV call is identified as somatic by the SNVQ subtraction method if (1) is called with high confidence from the tumor exome alignments, (2) is absent from the unfiltered SNVQ calls made from the normal exome, and (3) has coverage of at least 5 reads in the normal exome. In addition, SNVs called with high confidence from the tumor exome and are called with a different genotype from the normal exome are also identified as somatic by the SNVQ subtraction method.
The output of the CCCP pipeline includes the union of all somatic calls made by Strelka from Illumina and Ion Torrent technologies along with all calls made by SNVQ in at least one of the four runs on normal and tumor exomes generated using either technology (with high confidence in the case of tumor exome calls). For each variant position in this list, the read support for each of the four nucleotides in each of the input sequencing datasets (Illumina and Ion Torrent tumor and normal exomes and tumor RNA-Seq) are computed from alignments files in sequence alignment map (SAM) format using SAMtools (Li et al, 2009) and bedtools (Quinlan, 2014). The calls made for each of these positions by Strelka and SNVQ, their status in the Single Nucleotide Polymorphism Database (dbSNP), and the number of reads supporting each nucleotide are integrated into the CCCP output file. This enables rapid filtering of predicted SNVs based on a variety of criteria including variant caller and coverage support in the exome and RNA-Seq datasets generated by the two technologies, with no need to rerun CCCP.
2.1.2. Somatic SNV filter
By applying this filter, the user can select for validation candidate somatic SNVs with different levels of support from the available sequencing data. Using one of the three implemented filtering options is mandatory because the raw CCCP output includes (likely germline) SNVs detected in both the normal and tumor exomes as well as SNVs detected only in the normal exomes. All filters generate output in the same format as the CCCP tool, enabling the application of additional filters that take dbSNP status and RNA-Seq coverage into account and compatibility with the other downstream GeNeo tools.
The default option for the somatic SNV filter tool is to apply the filter called 2 Calls Cross-Platform (2CP) filtering. This method retains candidate SNVs that are called somatic at least twice, with concordant somatic alternative alleles, of the four possible somatic calls generated when running CCCP (Strelka Illumina, Strelka Ion Torrent, SNVQ Illumina, and SNVQ Ion Torrent). In addition, 2CP requires that the SNV be called as somatic at least once from data generated by each sequencing technology. The only exceptions are SNVs that have no coverage from one of the sequencing platforms, in which case the SNV is retained only when both Strelka and SNVQ make concordant somatic calls based on the sequencing reads generated by the other platform.
As previously reported in Sherafat et al (2020), 2CP offers an excellent balance between precision and recall, matching and often outperforming the F1-scores of both Strelka and the SNVQ subtraction method. However, the somatic SNV filter tool provides both less and more stringent filtering options that can be used when 2CP generates a number of candidates that is too low to generate sufficient candidate neoepitopes or too large for the multiplexing capacity of the AccessArray platform used for variant validation. The less stringent filter retains all SNVs called somatic by at least one of the two callers implemented in CCCP based on sequencing data generated by at least one of the two sequencing technologies.
Although the list of candidate somatic SNVs generated by the 1 Call filter is likely to include more false positives than the 2CP list, validating the calls by resequencing will detect and remove these false positives. The more stringent filter retains all SNVs that are called somatic at least three times of the four possible combinations of calling method and sequencing technology.
2.1.3. dbSNP filter
This tool takes as input a list of SNV calls in CCCP output format and removes from the list the variants that are annotated as “common” in the dbSNP database (Sherry et al, 2001). Such filtering can reduce the number of false positives caused by the misclassification of germline variants as somatic. However, particularly when variant validation is performed by targeted resequencing, applying dbSNP filtering should be avoided because, as noted by Koboldt (2020), a number of known recurrent cancer mutations have been included in dbSNP.
2.1.4. RNA-Seq coverage filter
This tool takes as input a list of SNV calls in CCCP output format and keeps only variants that have a minimum coverage (by default 1) of the alternative allele in the RNA-Seq reads. The RNA coverage of the alternative allele is calculated as the sum of coverages for the alternative allele in both the Illumina and Ion Torrent RNA-Seq reads. This filter can be used to prioritize variants with direct evidence of RNA expression among large numbers of somatic candidates. As the dbSNP filter, the RNA-Seq coverage filter must be used with caution because it may remove somatic SNVs with low levels of messenger RNA (mRNA) expression that may nevertheless generate MHC-I neoepitopes presented on the surface of tumor cells.
2.1.5. Variant calling format generator
The variant calling format (VCF) generator takes as input the file generated by CCCP (or one of the above filtering tools) and converts it to the standard VCF. The VCF is the required input of the Primer Designer tool described in the next section, and also allows the user to seamlessly integrate the output of CCCP with tools external to GeNeo if needed.
2.1.6. Summary report generator
This tool collects from the CCCP log statistics on the numbers of input reads, clean reads (reads passing the deduplication and QC steps), aligned reads, and coverage levels for the analyzed exome and RNA-Seq datasets.
2.2. Variant validation tools
Best practices for somatic variant calling from clinical NGS data require validation by an orthogonal technology (Koboldt, 2020). Sanger sequencing has long been considered the gold standard validation technique (Mu et al, 2016). However, Sanger sequencing has low throughput and suffers from low sensitivity for samples with low tumor content and/or subclonal somatic SNVs present in only a fraction of tumor cells. As an alternative, validation can be performed using the AccessArray nanofluidics system, which allows simultaneous PCR amplification of up to 480 candidate somatic SNVs from upto 48 samples, in conjunction with deep amplicon sequencing using either the Illumina or Ion Torrent platforms. GeNeo includes several bioinformatics tools designed to automate the validation process and check for possible sample mislabeling.
2.2.1. Primer designer
This tool takes as input a list of candidate somatic SNVs in VCF and attempts to design primers for variant validation using either Sanger sequencing or the AccessArray platform. Following recommended best practices, for Sanger sequencing, the tool attempts to design two nested primer pairs, one for PCR amplification and one for initiating Sanger sequencing on each strand. For the AccessArray platform primers are designed to target the manufacturer's recommended amplicon length and are tagged with universal forward/reverse common sequences CS1/CS2 for preparing sequencing libraries compatible with the Illumina or Ion Torrent platforms. Both Sanger and AccessArray primers can be designed for targeted amplification of the candidate somatic SNV positions from either genomic DNA or complementary DNA (cDNA).
2.2.2. AccessArray genotype caller
This tool demultiplexes AccessArray amplicon sequencing data from (typically replicated) tumor and normal samples and calls genotypes for each targeted SNV and each sample. It takes as input the AccessArray barcoded amplicon sequencing reads in fastq format and the set of targeted SNVs in VCF. The tool starts by creating an alternative genome reference with the reference allele substituted by the alternative allele at each targeted SNV position. A HISAT2 index is created for both the reference and the alternative genomes. To speed-up indexing and read alignment, the indices are built using only windows of ±500 bp around each targeted SNV. Next the tool demultiplexing the reads based on the AccessArray barcodes using the barcode splitter tool from the FASTX-Toolkit (FASTX-Toolkit, 2022). This generates multiple fastq files, one per AccessArray well.
The barcodes and sequencing adapters are then removed from the reads. Optionally duplicate reads can be removed using FastUniq (Xu et al, 2012), although this is necessary only for very low input samples such as single cells. Similar to CCCP, reads with average quality <20 are filtered out using Trimmomatic (Bolger et al, 2014). The remaining reads are then mapped to each one of the two references using HISAT2 (Kim et al, 2015), and the read alignments are used by SNVQ to call variants. SNVQ reports called SNVs as well as the coverage of each allele.
Using the alternative reference allows SNVQ to call genotypes and compute allelic fractions for positions where the AmpliSeq data does not support the alternative allele because sequencing data supporting a homozygous reference genotype will result in an SNV call when using the alternative reference. The first output of the tool is a tab-separated file that reports SNV genotype calls obtained by combining the two SNVQ runs, along with genotype quality and strand-specific coverages for both the reference and alternative alleles. The second output is a zipped file including the alignment files for each well in SAM format.
2.2.3. Alignment extractor
When validation via targeted resequencing is not possible (e.g., because appropriate primers cannot be designed or they fail to amplify the desired region) or is inconclusive, the current best practice is to perform a visual review of the read alignments supporting clinically relevant variants (Koboldt, 2020). The standard operating procedure for performing visual review (Barnell et al, 2019) requires using a genome browser like the integrative genomics viewer (IGV) (Robinson et al, 2017). This enables the identification of false positives caused by various artifacts including misalignment around indels or misalignments of reads from paralogous regions not represented in the reference genome (Koboldt, 2020). The alignment extractor tool helps streamline this process by generating a reduced reference genome that includes 500 bases before and after each SNV position of interest and generating BAM files that include only read alignments covering these positions. The tool accepts as input the target SNVs in VCF along with fastq files for the sequencing data that needs to be visualized, including any combination of matched tumor-normal exomes, RNA-Seq, or AccessArray targeted resequencing data generated using either the Illumina or Ion Torrent platforms (see the Galaxy interface in Fig. 4).
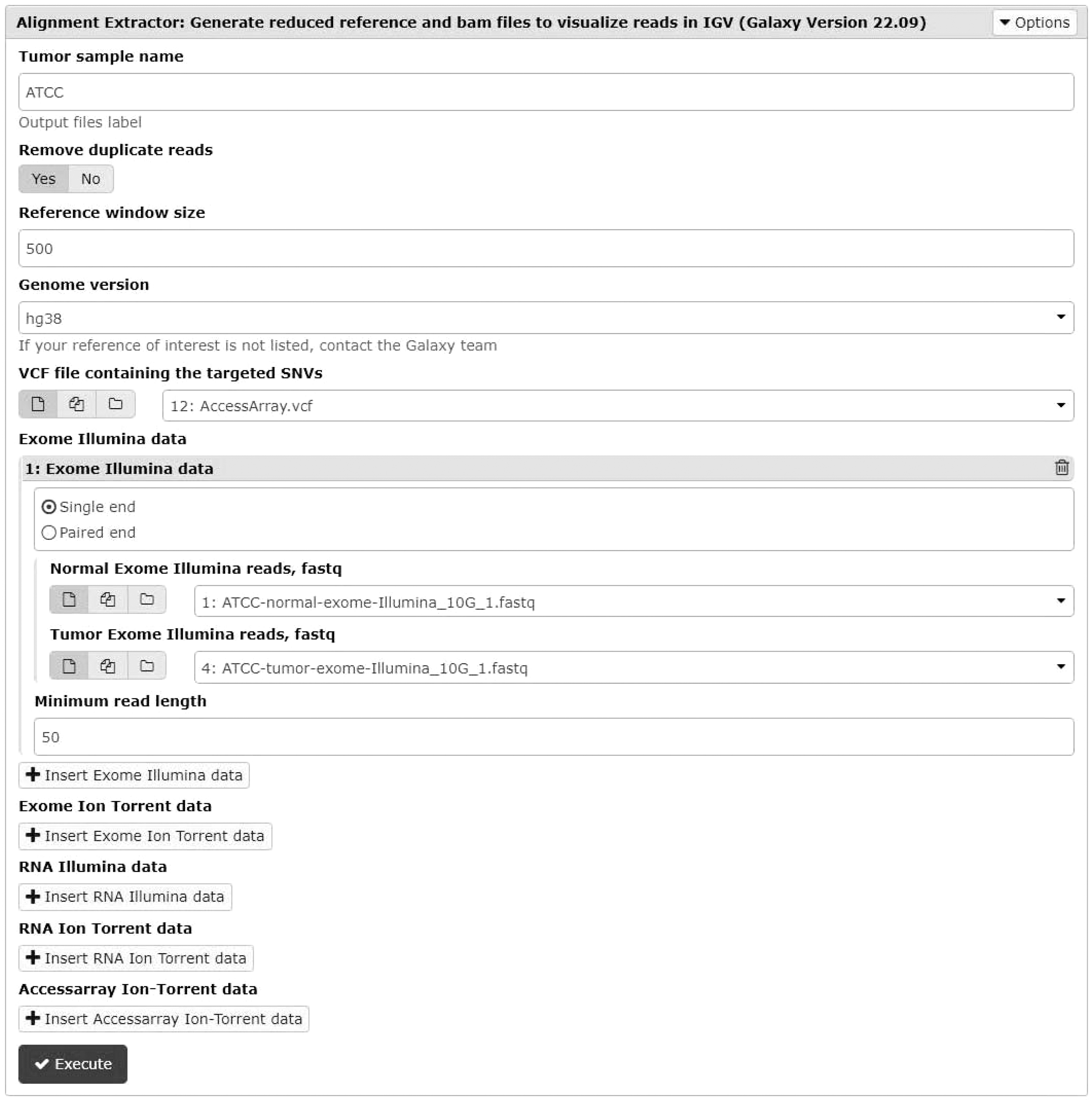
Galaxy interface for the alignment extractor tool.
2.2.4. MitoMatch
Sample contamination and mislabeling have been long-standing problems in biomedical research, and unfortunately, their prevalence has increased with the advent of high-throughput multi-omics datasets (Kho, 2021; Boja et al, 2018). Contaminated or mislabeled samples can result in a substantial increase in the number of false-positive somatic SNV calls, compromising the clinical utility of the sequencing data. To assist in the identification of contaminated or mislabeled samples, the MitoMatch tool can be used to infer the mitochondrial haplogroups for the sequenced exome and RNA samples. The tool accepts as input the same types of sequencing datasets accepted by the CCCP tool. The reads are mapped against the Reconstructed Sapiens Reference Sequence (RSRS) mitochondrial sequence and then SNVQ is used to call mitochondrial DNA (mtDNA) variants for each dataset. The Jaccard similarity-based algorithm in Alqahtani and Măndoiu (2020) is used to match the set of mtDNA variants called from each sample against a database of variants associated with the 2897 leaf haplogroups from Build 17 of PhyloTree (PhyloTree.org, 2016) as well as the two-haplogroup mixtures that can be formed using them. As shown in Alqahtani and Măndoiu (2020), this approach can quickly and accurately identify the PhyloTree haplogroup (or mixture of haplogroups) present in a sample, thus providing a simple method to simultaneously detect with high probability both sample contamination and sample mislabeling. †
2.3. Neoepitope prediction tools
Clinical use of personalized cancer vaccines requires predicting which mutated peptides, called neoepitopes, encoded by somatic cancer mutations get presented on the surface of tumor cells and are most likely to induce immune-mediated tumor control (reviewed in Lang et al, 2022). Most works in this area have focused on the prediction of neoepitopes presented on the surface of cancer cells by the MHC-I molecules, which can mediate the cytotoxic effects of CD8+ T cells. In this section, we describe the GeNeo tools designed to assist with neoepitope prediction and prioritization.
2.3.1. Seq2HLA
The MHC-I molecules are highly polymorphic, with three gene loci and thousands of alleles cataloged in the human population. Each individual carries up to 6 distinct alleles, one maternal and one paternal allele for each of the three loci. Because the repertoire of mutated peptides presented on the surface of cancer cells depends on the combination of alleles they express, the Seq2HLA tool uses the algorithm in Boegel et al (2013) to infer the MHC-I alleles from Illumina paired-end tumor RNA-Seq reads provided as input in fastq format. In addition, Seq2HLA also infers MHC class II alleles and the expression level for each MHC locus.
2.3.2. Neopitope finder
This tool uses NetMHC 4.0 (Andreatta and Nielsen, 2016) to predict neoepitopes encoded by somatic SNVs given as input in CCCP format. Using CCDS annotations (Pruitt et al, 2009), the tool extracts, for each input SNV, one or more cDNA sequences of 63 nucleotides (or shorter depending on the location of the SNV within the transcript) with the SNV in the middle. It then translates the cDNA into peptide sequences of 21 amino acids or shorter. If the SNV is nonsynonymous resulting in different wild-type and mutant long peptides, the tool calls NetMHC to compute the binding affinity (half maximal inhibitory concentration or IC50) of each 8, 9, 10, and 11-mer peptide containing the mutated amino acid and their corresponding wild-type peptides (Fig. 5). The tool also computes for each mutated peptide the DAI previously introduced by us in Duan et al (2014).
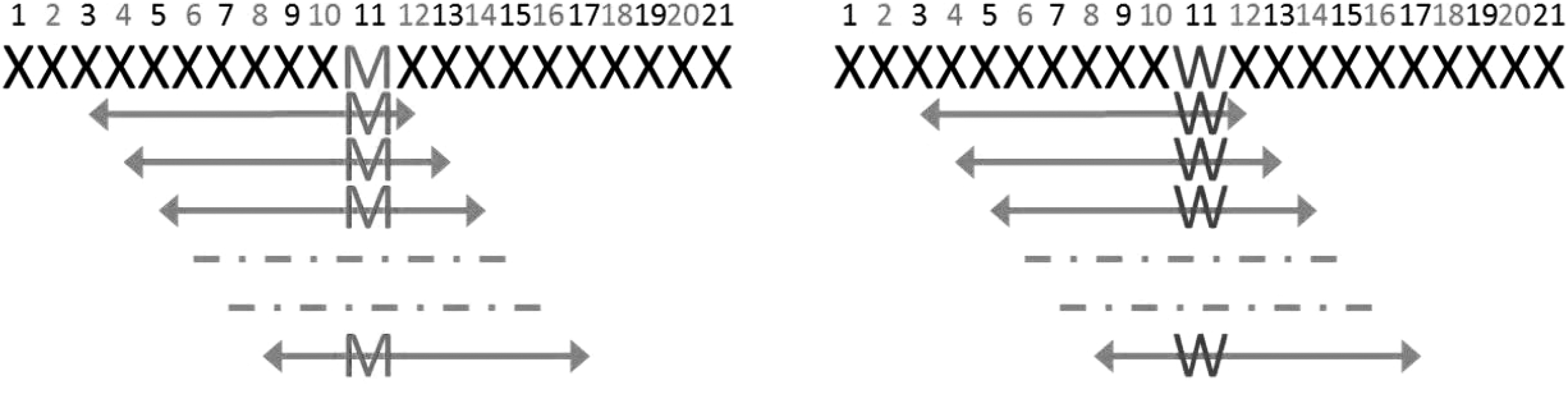
Diagram illustrating the generation of all possible mutant and corresponding wild type 8-mer peptides spanning a somatic SNV position. The same strategy is used for generating 9, 10, and 11-mer mutant and wild-type peptides before passing them NetMHC for IC50 prediction.
The DAI was originally defined in Duan et al (2014) as the logarithm of the ratio between the likelihood of the mutant and wild-type k-mers under a profile weight matrix (PWM) model trained for the MHC-I allele by NetMHC 3.0. Because newer versions of NetMHC and other prediction programs including NetMHCpan no longer include a PWM model and instead use neural networks to directly predict IC50, the Neoepitope Finder tool uses a DAI approximation computed as the difference between the base 2 logarithms of the wild-type k-mer IC50 and the mutant k-mer IC50. In addition to the main output file, which includes all possible 8, 9, 10, and 11-mer neoepitopes along with their mutant and wild-type IC50 and DAI scores, the neoepitope finder tool also outputs a summary file including a list of the input SNVs characterized as missense, nonsense, nonstop, or nonsynonymous. It also includes overall counts for each SNV type (Fig. 6).

The tail of a sample SNV statistics file generated as a secondary output of the Epitope Finder tool. HT, heterozygous.
2.3.3. IC50 and DAI filters
Prioritizing neoepitopes based on their potential to mediate tumor control or rejection is currently a very active area of research, including consortium efforts such as Wells et al (2020). Most commonly neoepitopes are prioritized based on their predicted binding affinity (usually measured as the IC50). However, immunization experiments in mouse models (Brennick et al, 2021; Duan et al, 2014) and retrospective analyses of clinical outcomes of TCGA patients (Ghorani et al, 2018; Rech et al, 2018) suggest that neoepitopes with high DAI may be more important for long-lasting tumor control or rejection. GeNeo includes two filtering tools, which allow selecting from the raw output generated by the neoepitope finding tool the mutant peptides with IC50 values below a user-specified threshold (by default 500 nm), or DAI scores above a specified threshold (by default 1). If desired the two filters can be combined to generate lists of candidates that meet both maximum IC50 and minimum DAI criteria.
2.4 Ethics approval and consent to participate
The sequencing data analyzed in this manuscript was generated as part of the Phase I clinical trial “Study of Oncoimmunome for the Treatment of Stage III/IV Ovarian Carcinoma” (ClinicalTrials.gov Identifier: NCT02933073), conducted in accordance with FDA and Human Subject Protection regulations under IRB approval from the UConn Health Center. Written informed consent was obtained from all subjects.
3. Ge Neo APPLICATION IN A PHASE I CLINICAL TRIAL
The clinical trial “Study of Oncoimmunome for the Treatment of Stage III/IV Ovarian Carcinoma” (ClinicalTrials.gov Identifier: NCT02933073) was established to test the safety and feasibility of the personalized OncoImmunome vaccine in stage III/IV ovarian carcinoma patients. OncoImmunome is a tumor-specific vaccine consisting of a mixture of 7–10 synthetic long peptides each containing 17–18 amino acids and encoding neoepitopes predicted to elicit anti-tumor immune responses. Women with stage III/IV ovarian cancer who have received the standard of care treatment for their cancer and were in clinical remission were given this vaccine, which is produced specifically for each participant based on somatic mutations identified from NGS sequencing data.
3.1. Exome, RNA, and AccessArray amplicon sequencing
Surgically resected tumor tissue samples were flash-frozen in LN2. Genomic DNA (gDNA) and mRNA from the tumor samples were purified using AllPrep DNA/RNA Mini Kit (Qiagen). Blood samples were collected using ACD Vacutainer blood collection tubes. gDNA was extracted using DNeasy Blood and Tissue Kit (Qiagen). The exome and transcriptome of these samples were sequenced using Illumina and Ion Torrent sequencing platforms.
Illumina sequencing
gDNA samples were prepared for exome sequencing using the Nextera Rapid Capture Exome kit (Illumina) and sequenced on the Illumina NextSeq 500 System using Mid Output v2 300 cycle kit. The RNA sample was prepared for RNA sequencing using the Illumina Stranded mRNA Library Prep kit, Set A, and sequenced on the Illumina NextSeq 500 using Mid Output v2 300 cycle kit.
Ion Torrent sequencing
Whole-exome sequencing of the isolated gDNA was performed using Ion AmpliSeq Exome RDY kit (ThermoFisher Scientific) on the Ion Torrent Proton or S5 sequencers. RNA libraries were prepared using the Ion Total RNA–Seq kit v2 from Life Technologies and also sequenced on Ion Torrent Proton or S5 sequencers.
AccessArray sequencing
The Fluidigm AccessArray instrument was used to amplify candidate somatic SNVs and make sequencing libraries from bulk gDNA from flash-frozen tumor samples using pairs of primers designed using the GeNeo primer designer tool. The AccessArray library was then sequenced using an Ion Torrent Personal Genome Machine (PGM).
3.2. Somatic SNV calling and neoepitope prediction using GeNeo
The CCCP tool was used to call somatic SNVs using Illumina and Ion Torrent exome and RNA sequencing data from tumor tissue and blood (matching normal) samples collected for four patients. Sequencing and read mapping statistics are given in Table 1. The output of CCCP was filtered using the 2CP filter. The resulting candidate somatic SNVs were validated using AccessArray targeted sequencing performed on quadruplicate bulk tumor and normal samples using primers designed using the GeNeo primer design tool. The AccessArray genotype caller tool was applied to demultiplex the AccessArray sequencing reads and call genotypes for candidate SNVs in each replicate sample. Next, we used our Neopitope Finder tool to generate the mutant peptide sequences, compute IC50 values for both mutant and corresponding wild type peptides, and calculate DAI scores.
Sequencing Reads Statistics for Four Ovarian Carcinoma Patients
For each patient, the table shows the raw and aligned read counts and the average exome coverage based on normal exome, tumor exome, and tumor RNA-Seq reads generated using Illumina and Ion Torrent platforms.
Statistics collected for various stages of GeNeo processing, including the number of somatic SNVs identified by 2CP and the number of SNVs predicted to yield neoepitopes at different IC50 and DAI thresholds, are given in Table 2. For comparison, the number of somatic SNV calls predicted by the less stringent “1 Call” filter is also included. The results show that somatic SNVs predicted by 2CP have a consistently high validation rate, typically yielding sufficient putative neoepitopes for inclusion in the cancer vaccine. For a larger set of neoepitope choices the set of SNVs resequenced on the AccessArray can be expanded to include more candidates from the “1 Call” list. Because the full “1 Call” list is typically too large to fit on a single AccessArray IFC (which has multiplexing capacity limited to 480 SNVs), semi-supervised learning can be used to select from the “1 Call” SNVs a subset of 480 candidates that are most likely to be somatic (Sherafat et al, 2020).
Somatic Single Nucleotide Variant and Predicted Neoepitope Statistics for Four Ovarian Carcinoma Patients
CCCP 1Call and CCCP 2CP columns show the number of somatic SNVs that were called by CCCP using the respective filters. HT&NonSyn shows the number and percentage of the 2CP somatic calls that are both heterozygous in tumor samples and nonsynonymous. Validation results based on targeted resequencing using the AccessArray and Ion Torrent PGM sequencing are also included. The Designed Primers columns show the number and percentage of the HT&NonSyn SNVs for which primer design was successful. Working Primers shows the number and percentage of designed primers that generated sequencing reads. Validated shows the number and percentage of SNVs with working primers that were actually validated as somatic. The last four columns show the number of validated SNVs predicted to yield neoepitopes based on commonly used IC50 and DAI thresholds.
2CP, 2 Calls Cross-Platform; CCCP, Consensus Caller Cross-Platform; DAI, Differential Agretopic Index; IC50, half maximal inhibitory concentration.
4. CONCLUSION
For simplicity and ease of use, the GeNeo toolbox is available via a public Galaxy portal available at https://neo.engr.uconn.edu/. A virtual machine image for running GeNeo on user's premises is available for academic use upon request. The image runs a Galaxy server configured with all GeNeo tools, including their dependencies, genome references, and read mapping indices. The only exception is NetMHC, which has to be downloaded directly from https://services.healthtech.dtu.dk/services/NetMHC-4.0/ and installed by the user because distribution to third parties is prohibited under the terms of the academic license agreement.
Footnotes
AUTHOR DISCLOSURE STATEMENT
PKS and IIM are inventors of patent US-10501801, “Identification of tumor-protective epitopes for the treatment of cancers”, disclosing the use of DAI for predicting whether immunization with a particular epitope will be protective against the tumor. The other authors declare they have no conflicting financial interests.
FUNDING INFORMATION
This work was supported in part by the Neag Cancer Immunology Translational Research Fund and the Eversource Energy Chair in Experimental Oncology. ES was supported in part by a US Department of Education GAANN fellowships (Office of Postsecondary Education Grant No. P200A180092). IIM was supported in part by the US National Science Foundation Award 1564936. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.