
Abstract
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused a serious threat to public health and prompted researchers to find anti-coronavirus 2019 (COVID-19) compounds. In this study, the long short-term memory-based recurrent neural network was used to generate new inhibitors for the coronavirus. First, the model was trained to generate drug compounds in the form of valid simplified molecular-input line-entry system strings. Then, the structures of COVID-19 main protease inhibitors were applied to fine-tune the model. After fine-tuning, the network could generate new molecular structures as novel SARS-CoV-2 main protease inhibitors. Molecular docking exhibited that some generated compounds have the proper affinity to the active site of the protease. Molecular Dynamics simulations explored binding free energies of the compounds over simulation trajectories. In addition, in silico absorption, distribution, metabolism, and excretion studies showed that some novel compounds could be formulated as orally active agents. Based on molecular docking and molecular dynamics simulation studies, compound AADH possessed significant binding affinity and presumably inhibition against the SARS-CoV-2 main protease enzyme. Therefore, the proposed deep learning-based model was capable of generating promising anti-COVID-19 drugs.
INTRODUCTION
In December 2019
The SARS-CoV-2 main protease enzyme (3 C-like protease) is a key CoV target that plays a critical role in the SARS-CoV-2 life cycle as this enzyme influences the process of viral replication and transcription (Jin et al., 2020). The main protease is a quintessential enzyme that contributes significantly to the life cycle of SARS-CoV-2, and inhibition of the enzyme activity would block viral replication. Since no human proteases with a similarly specified cleavage are characterized, the potential inhibitors are unlikely to be toxic. The enzyme consists of an asymmetric unit, including 305 amino acid residues with Cys145 and His41 catalytic dyads in the active site (Zhang et al., 2020). Experimental observations demonstrated the half maximal effective concentration value of 16.77 μM for the N3-Michael acceptor inhibitor which exhibited very potent antiviral effects against the main protease enzyme (Jin et al., 2020). Herein, we have used SARS-CoV-2 main protease as a target enzyme to identify the potent antiviral compounds with potential inhibitory activities against the enzyme.
Since traditional methods for drug discovery are costly and time-consuming processes that sometimes take decades to complete, the application of artificial intelligence techniques in drug discovery has achieved great popularity and success. Accordingly, Computer-aided drug design techniques were applied to introduce novel effective anti-COVID agents (Karade and Karade, 2022).
Deep learning is a subset of machine learning that uses neural networks with multiple layers of nonlinear processing units to model the high-level abstractions in the data. In recent years, the applications of deep learning in pharmaceutical research, including virtual screening, absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties, production of new chemical structures (Bilodeau et al., 2022; Mukherjee et al., 2022), and quantitative structure activity relationship (QSAR) models are increasing (Wang et al., 2022). Recurrent neural network (RNN) is one of the most widely used representative architectures in deep learning. Unlike other artificial neural networks, RNN is especially useful when working with sequence data in which the current output depends on previous inputs (Eshaghnezhad et al., 2022). It has been used in many fields of research, such as natural language processing (Veliz-Cuba et al., 2015), translation (Wu et al., 2016), music composing (Subakan and Smaragdis, 2017), and drug design (Gupta et al., 2018).
The simplified molecular-input line-entry system (SMILES) is a specification in the form of a line notation, which describes chemical structures using short ASCII strings. The probability distribution of the next character in a SMILES string can be predicted by a pretrained RNN model. RNN can be used as a generative model for molecular structures by learning the possible distribution of characters in a SMILES string. Therefore, this type of deep neural network can be used to generate new drug compounds. Compared to traditional ligand-based methods, the RNN-based generative model only requires SMILES information of chemical ligands to train the model and can generate a large set of new and potential structures. The unique features of the RNN make it very useful in de novo drug design, as well as virtual screening.
Yuan et al. (2017) conducted their studies using RNN to generate new chemical structures. After training the model on a number of SMILES strings, the RNN based model was surprisingly able to generate valid new SMILES strings that did not exist in the training set. Segler et al. (2018) used canonicalized SMILES representation to train the RNN. They also showed how the model training using a focused set of actives toward a biological target could create a fine-tuned model capable of producing a high fraction of predicted actives. Researchers in Olivecrona et al. (2017) used the concept of reinforcement learning to increase the efficiency of SMILES-generating RNNs. In this method, the parameters of the model were optimized to generate strings with a high evaluation score. They used this approach to produce sets with low sulfur content and high predicted target activity.
Long short-term memory (LSTM) and gated recurrent unit (GRU) networks are two main types of RNNs capable of learning order dependence in sequence prediction problems (Bouziane and Khadir, 2022). Shi et al. (2020) used a GRU-based network to create a molecular generative model of ADAM10 inhibitors. The results showed that the GRU-based model could accurately learn SMILES grammars from molecules and was able to generate potential new ADAM10 inhibitors.
In this article an LSTM-based RNN was used to create a model that generates anti-SARS-CoV-2 small molecules. In the first stage, the model was trained to produce a library of valid SMILES disciplines with high accuracy. Then, the compounds with inhibitory activity against the main protease enzyme of the coronavirus were used to fine-tune the model. Finally, docking studies and molecular dynamic (MD) simulations were performed on the selected compounds in complex with the main protease enzyme to probe the binding affinities and binding free energies of the compounds upon complexation, respectively.
The rest of this article is organized as follows. Section 2 provides essential materials and introduces the proposed method in detail. Section 3 gives an explanation of the experimental study conducted in this work. We conclude the article in Section 4.
MATERIALS AND METHODS
Proposed method
Our proposed method generates drug compounds as SARS-CoV-2 main protease inhibitors by training an LSTM-based neural network. The proposed method has a two-stage learning mechanism. In the first stage, the model learns to produce chemical structures, and by further training in the second stage, the model learns to generate molecular structures like our target compounds. Figure 1 shows the general steps of the proposed method.
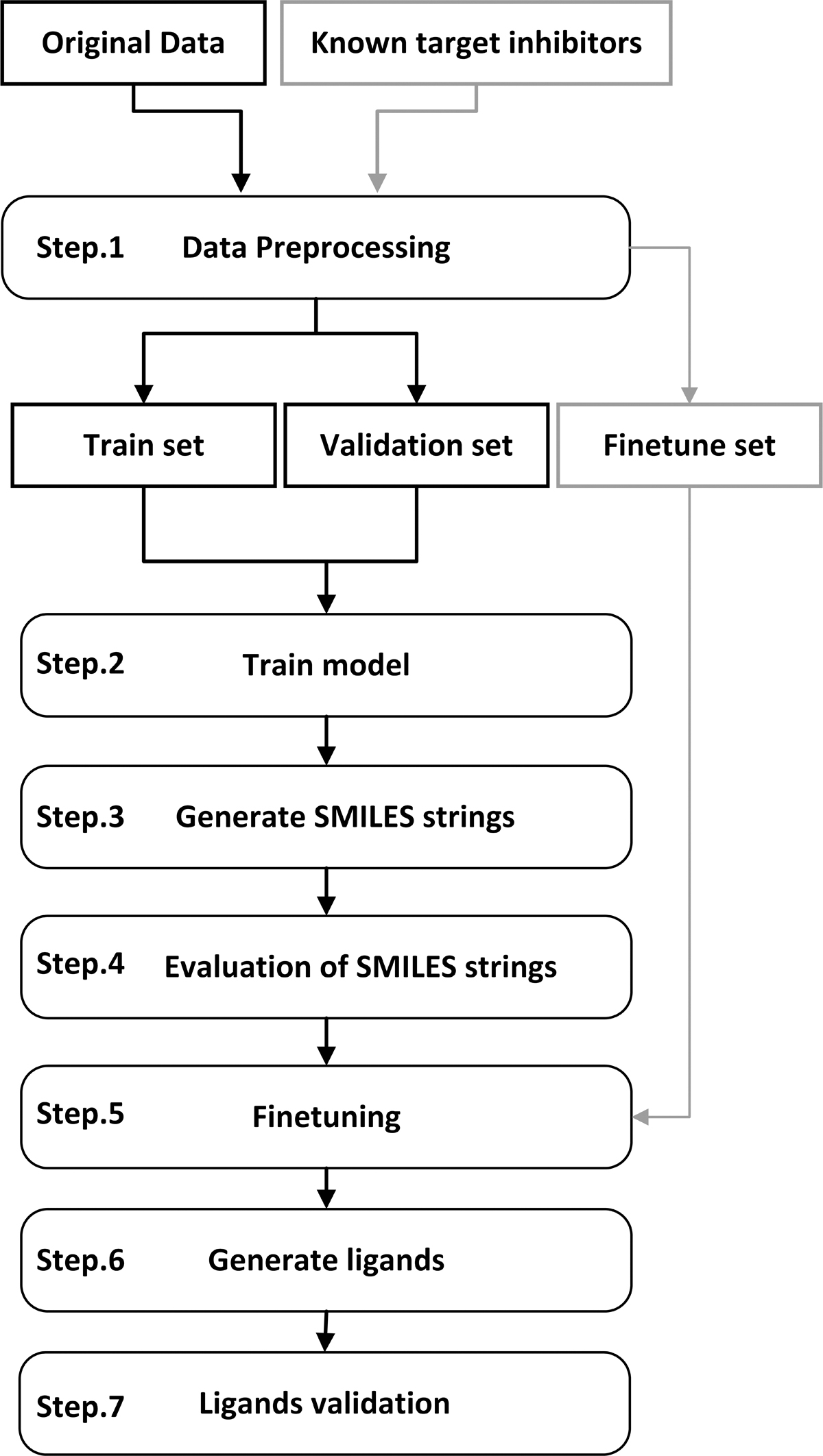
The flow chart of the proposed method. SMILES, simplified molecular-input line-entry system.
According to Figure 1, the proposed methodology includes seven steps. In the first step, the structures needed for model training were selected according to Section 2.2 and then were tokenized. The output of step 1 consists of three datasets that include training, validation, and fine-tuning structures. Then, in the next step, the proposed model was trained according to Section 2.4. In the third step, some structures were generated to evaluate the model's ability to produce valid SMILES structures. After the evaluation step, we trained the model again with the SARS-CoV-2 main protease inhibitors. In step 6, a set of potential novel inhibitor structures were generated. Finally, in step 7, the novel inhibitors were evaluated through Molecular docking, ADME studies, and Molecular Dynamics simulations (The details are described in Section 3).
In this study, annotated nanomolar activities from the ChEMBL dataset (Gaulton et al., 2017) were selected and combined with MOSES dataset (Polykovskiy et al., 2020) which is based on the ZINC dataset (Sterling and Irwin, 2015). The dataset resulting from the combination contained 4,591,276 molecules in total. In the following, molecules with molecular weight (MW) ranging from 250 to 350 Daltons, rotatable bonds not >7, and XlogP ≤3.5 were selected from the dataset. Then, molecules containing charged atoms or atoms besides C, N, S, O, F, Cl, Br, and H or cycles longer than eight atoms were removed. Finally, preprocessing was done to remove duplicates, salts, and stereochemical information. At the end of the process, we have 2,492,858 SMILES strings prepared to use as a train set. Figure 2 shows the preparation steps of the training dataset schematically. The filtering and standardization steps were implemented using the RDKit library. The dataset consisting of 500 molecules screened against SARS-CoV-2 main protease was used for fine-tuning. Molecules in the fine-tuning dataset were submitted to the same preprocessing protocol described above.

Training dataset.
Before describing the architecture of the applied model, we explain the structure of the input data. Molecules in the form of SMILES strings were used as the model input. SMILES is a widely used notation in drug design and cheminformatics to represent chemical structures as text strings. It allows for compact and unambiguous representation of molecular structures, making it easy to store, exchange, and process chemical information. SMILES strings are very concise compared to other chemical file formats like MOL or SDF. This compactness makes them efficient for storing and exchanging large databases of chemical structures, which is crucial in drug discovery and virtual screening.
The SMILES strings were tokenized before processing. Token G (Go) and token E (End) were added to the structure of the molecules to indicate the beginning and end of the SMILES, respectively. Given that the length of the input SMILES strings varies between 34 and 74 tokens, a number of A tokens have been used as padding after the E token to equalize the size of all inputs. After performing the preprocessing operations, the structures in training dataset were converted to a sequence of tokens with a fixed length of 74. Figure 3 shows the structure of SMILES strings after preprocessing.
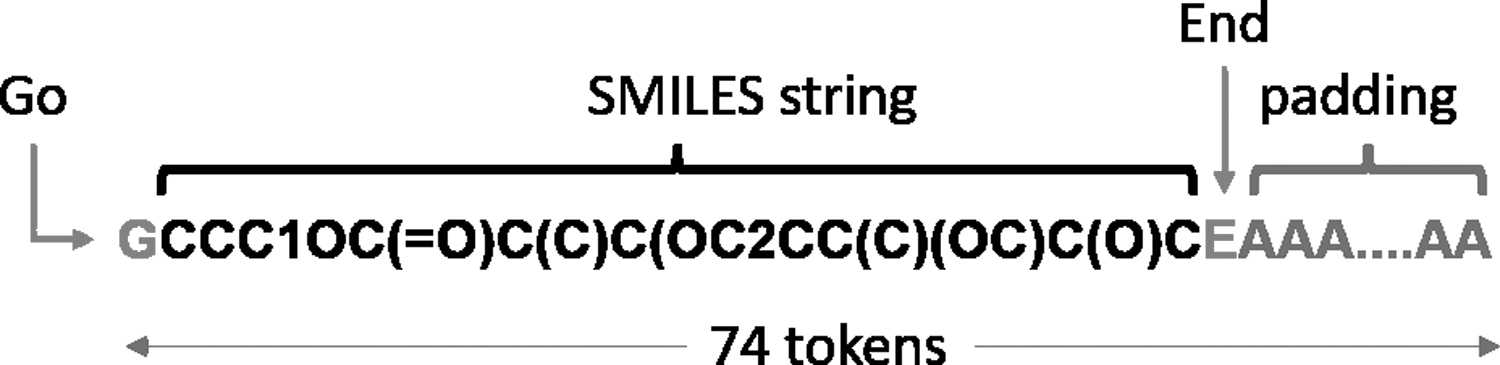
A sample SMILES string.
RNNs suffer from the matter of vanishing gradients. The gradients carry information utilized in the RNN, and when the gradient becomes too small, the parameter updates become insignificant. This makes the training of long data sequences difficult (Pascanu et al., 2013). LSTM is a special kind of RNNs, capable of learning long-term dependencies. A common unit of LSTM is composed of a cell, an input gate, an output gate, and a forget gate. The cell remembers values over arbitrary time intervals, and the three gates regulate the flow of information into and out of the cell. Input gate is used to quantify the importance of the new information carried by the input. The output gate determines the value of the next hidden state. This state contains information on previous inputs. A Forget Gate responsibility is to decide whether we should keep the information from the previous timestamp or forget it (Bouziane and Khadir, 2022).
The architecture of the model used in this research is shown in Figure 4. This architecture consists of two LSTM layers (Hochreiter and Urgen Schmidhuber, n.d.), in which their outputs were connected to a dense layer. After this layer, there is a neuron with a softmax activation function. The SMILES string data in each step were processed by a set of gates, including input gate, output gate, and forget gate. Then, important information was passed to the next cell. LSTM allows us to control what information was passed by the hidden state to the next cell and what was not considered during data processing. In LSTMs, due to the existence of the cell state, the model can have important information in memory for a longer time. This increases the efficiency of the model in processing a long sequence of data. The proposed model processes each token of the input SMILES string at each step and generates a probability distribution of all possible tokens to discover the rules in the structure of the SMILES. This probability distribution was processed by the softmax function and predicts the next step token. In the proposed model, SMILES strings were generated token by token. It will be explained in more detail in the next section.

The architecture of the generative model. LSTM, long short-term memory.
To train the model, first, 2,492,858 SMILES were preprocessed as described in Section 2.2 and were given to the model in the format of Figure 3. The model predicts the next token for each input token and by comparing it with the input token in the same position measures the accuracy of its prediction. Thus, it discovers the rules for producing SMILES string structures. To train the RNN model, the maximum likelihood estimation method has been used. In this method, the goal of the model was to maximize the probability assigned to the next correct token for each vector in the array. In this step, the token G, which was considered as the starting word of the sequence, was fed to the model and the model generates the next token. Then, to generate a complete sequence, the generated token was fed as input to the model to generate the next token. This continues until we reach the token E, which represents the end of the sequence. During the token production process, the output of each phase was concatenated to the output of the previous phase. In each position of the SMILES strings, the amount of categorical cross-entropy error between the next token was predicted and calculated. We denote the mean value of this error by L, which was calculated for all tokens among all molecules by Eq. (1).

After training, the model can generate structures similar to the input data. The model used in this study was similar to the model proposed in Gupta et al. (2018). However, changes have been made to the input data and the number of model epochs, resulting in better performance than the original model. Details of this comparison are given in Section 3. By creating an efficient model for the production of drug structures, this model can be used to produce drug structures with a specific purpose. This will be done by fine-tuning the model which will be explained in the following.
Fine-tuning
In this study we fine-tune the model as a main part of transfer learning which is a machine learning technique that leverages knowledge gained from solving one task and applies it to a different but related task. In transfer learning, a pretrained model, typically trained on a large dataset, is used as a starting point for a new task, allowing the model to benefit from the learned features and representations. The process of transfer learning involves two main steps of pretraining and fine-tuning.
Deep learning models often require a large amount of labeled data to achieve good performance. However, collecting and annotating large datasets can be time-consuming and expensive. Fine-tuning is a popular technique in deep learning which refers to the process of taking a pretrained neural network model and further training it on a new dataset. Instead of training a model from scratch, fine-tuning allows us to leverage the knowledge and learned representations from a pretrained model. Initially, a deep learning model, such as LSTM in our work, is trained on a large dataset, often referred to as the pretraining dataset. This dataset is usually diverse and generic. Once the model is trained on the pretraining dataset, it learns useful features and representations that capture general patterns and knowledge from the data. These learned features can be valuable for related tasks. To fine-tune the model for a specific task, you need a smaller dataset that is specific to the task at hand. This approach is particularly useful when the new dataset is small or when you don't have sufficient computational resources to train a model from scratch.
Since we had a limited number of SARS-CoV-2 inhibitor structures, we used the transfer learning technique. After the training of the model using a pretraining dataset which is described in Section 2.1, the model can produce valid, varied, and new SMILES strings. However, to produce inhibitor structures, it was necessary to further train the model with similar structures. This further train helps the model to update its weights based on the loss of the model during learning in such a way that it should generate similar structures to the presented structures. To fine-tune the network, 22 inhibitors of SARS-CoV-2 main protease and its co-crystallized ligand have been used. Detailed information on the inhibitors is given in a Supplementary Data. All these structures have been selected according to the results of previous studies (Tian et al., 2021). At this stage, 100 inhibitor structures have been generated, and we evaluated them to select effective inhibitors.
Molecular docking analysis of main protease inhibitors
The 2.6 Å resolution X-ray crystallographic structure of SARS-CoV-2 main protease in complex with its co-crystallized peptide-like inhibitor (N3) was retrieved from the RCSB Protein Data Bank (PDB ID: 6LU7; Jin et al., 2020). The structure of the enzyme was preprocessed, minimized, and refined using the Protein Preparation Wizard implemented in Maestro 11.6 module (Schrodinger Release 2020-Maestro, S., LLC, New York, NY, USA) implemented in the Schrodinger suite (Madhavi Sastry et al., 2013). This step involved eliminating crystallographic waters, missing hydrogens/side chain atoms were added, and the appropriate charge and protonation state was assigned to the receptor structure corresponding to pH 7.0 considering the appropriate ionization states for the acidic, as well as basic, amino acid residues. The structure was subjected to energy minimization using the Optimized Potentials for Liquid Simulations-2005 (OPLS-2005) force-field * (Harder et al., 2016) with a root mean square deviation (RMSD) cutoff value of 0.30 Å to relieve the steric clashes among the residues due to the addition of hydrogen atoms.
The preparation of the ligands, namely, N3-inhibitor and the studied compounds, was performed using the LigPrep (Schrodinger Release 2020-1: LigPrep, S., LLC, New York, NY, USA) module of Schrodinger Suite which performs the addition of hydrogens, adjusting realistic bond lengths and angles, correct chiralities, ionization states, tautomers, stereo chemistries, and ring conformations. Partial charges were assigned to the structures using the OPLS-2005 force-field, and the resulting structures were subjected to energy minimization until their average RMSD reached 0.001 Å. The ionization state was set at the neutral pH using the Epik ionization tool (Schrodinger Release 2020-1: Epik, S., LLC, New York, NY.).
Molecular-docking-based virtual screening was performed using the Glide workflow of Maestro 11.6 (Friesner et al., 2004) to identify suitable compounds that strongly bind to the main protease enzyme. The receptor grid was generated as center coordinates (X = −10.81, Y = 12.41, Z = 68.93) using two cubical boxes having a common centroid to organize the calculations: a larger enclosing and a smaller binding box with dimensions of 24 × 24 × 24 Å and 32 × 32 × 32 Å, respectively. The grid box is centered on the centroid of the ligands in the complex, which was sufficiently large to explore a larger region of the enzyme structure. The ligands were docked using the “Extra-Precision” mode (XP), and the best poses were selected based on XP G-scores.
ADME properties
The ADME properties of the novel structures were predicted using the Swiss ADME online property calculator (www.swissadme.ch; Daina et al., 2017). Topological polar surface area, number of rotatable bonds, MW, the logarithm of the partition coefficient (miLog P), number of hydrogen bond acceptors (n-ON), number of hydrogen bond donors (n-OHNH), Lipinski's rule of five criteria, and finally gastrointestinal (GI) absorption were calculated (Lipinski et al., 2001). Furthermore, the toxicity risks of the compounds, including mutagenic, tumorigenic, and irritant, were evaluated by organic chemistry portal.
MD simulations
The ff14SB (Ponder et al., 2003; Salomon-Ferrer et al., 2013a,b) and the general AMBER force fields (Wang and Wolf, 2004) were used to parameterize the enzyme and the considered ligands using Leap implemented in Amber 20 (Case et al., 2020). The system was solvated using the TIP3P (Harrach and Drossel, 2014) explicit water in a cubic box with a 10 Å distance around the system, and Na þ ions were added randomly to neutralize the complex. The partial Mesh Ewald (Harvey and De Fabritiis, 2009) method was used to account for the long-range electrostatic forces using a cutoff of 12 Å, and the Secure Hash Algorithm Keccak (SHAKE) algorithm (Ryckaert et al., 1977) was used to constrain all the hydrogen atom bonds. Energy minimizations were performed in two stages with 2500 steps of steepest decent minimization followed by 2500 of the conjugated gradient to remove the bad contacts. The first stage was followed with a harmonic restraint of 500 kcal mol−1 on the solute molecule, whereas ions and water molecules were relaxed. In the second stage of minimization, the restraints were removed and the whole system was relaxed. The minimized system was gradually heated up from 0 to 300 K with a weak harmonic restraint of 10 kcal mol−1 A−2 to keep the solute fixed for 200 ps. The 50 ps density equilibration with weak restraints followed by the 500 ps constant pressure equilibration at 300 K was performed. Finally, the 120 ns production of MD simulations using the Graphics Processing Unit accelerated version of Partial Mesh Ewald Molecular Dynamics (Srinivasan et al., 1998; Tian et al., 2019; Wang et al., 2019) was run at a constant temperature of 300 K and constant pressure at 1 atm.
Postdynamics analysis and binding free energy calculation
The 120 ns MD trajectories were analyzed to calculate the RMSD, root mean square fluctuation (RMSF), the radius of gyration (Rg), and intermolecular hydrogen bonding interactions using the CPPTRAJ module (Roe, 2013) implemented in Amber20 as the main program for processing coordinate trajectories and data files.
The molecular Mechanics/Generalized Born Surface Area (MM-GBSA) binding free energy method [28, 29] was applied to calculate the relative binding free energies. All water molecules and counter ions were stripped using the CPPTRAJ module. The binding free energy (ΔGbind) was calculated with the MM-GBSA method for each system using the following equations:
The gas phase energy (ΔEgas; Eq. 4) is the sum of the internal (Eint; Eq. 5), van der Waals (EvdW), and Coulombic (Eelec) energies. The solvation free energy is the combination of polar (GGB) and nonpolar (Gnonpolar, solvation) contributions (Eq. 6). It is notable that the GGB polar solvation is calculated using the Generalized Born solvation model with the dielectric constant 1 for solute and 80.0 for the solvent (Onufriev et al., 2000). The nonpolar free energy contribution was calculated using Eq. (7), where the surface tension proportionality constant, γ, and the free energy of nonpolar solvation of a point solute, β, were set to 0.00542 kcal mol−1 A−2 and 0 kcal mol−1, respectively (Gohlke et al., 2003). The solvent-accessible surface area was calculated by the linear combination of the pairwise overlap method (Weiser et al., 1999).
Performance of generative model
Before the fine-tuning stage, to evaluate the efficiency of the proposed model in the production of drug structures, 10,000 drug structures were produced and three evaluation criteria of validity, uniqueness, and novelty were used. These three criteria were defined as follows:
Validity: performance of the model in capturing explicit chemical constraints. A SMILES string is considered valid if it can be parsed by RDKit without errors. Uniqueness: percentage of unique canonical SMILES generated. This criterion checks that the model does not collapse for producing only a few typical molecules. Novelty: percentage of valid molecules in generated data that are not present in the training dataset.
The evaluation results showed that the model could generate valid structures with a value of 96.56%. This value is 92% (Gupta et al., 2018), which has a similar architecture to our model. In addition, the uniqueness and originality of the model are 99.68% and 86.74%, respectively.
The loss value of the model was calculated through Eq. (1). Figure 5 shows the model loss value in the training and validation steps in 40 different epochs. The training loss indicates how well the model is fitting the training data, while the validation loss indicates how well the model fits new data. As Figure 5 shows, the training and validation loss both decrease over time, and the gap between them is small. Therefore, we can conclude that the training process is done well, and we have a good fit without the overfitting or underfitting problems.
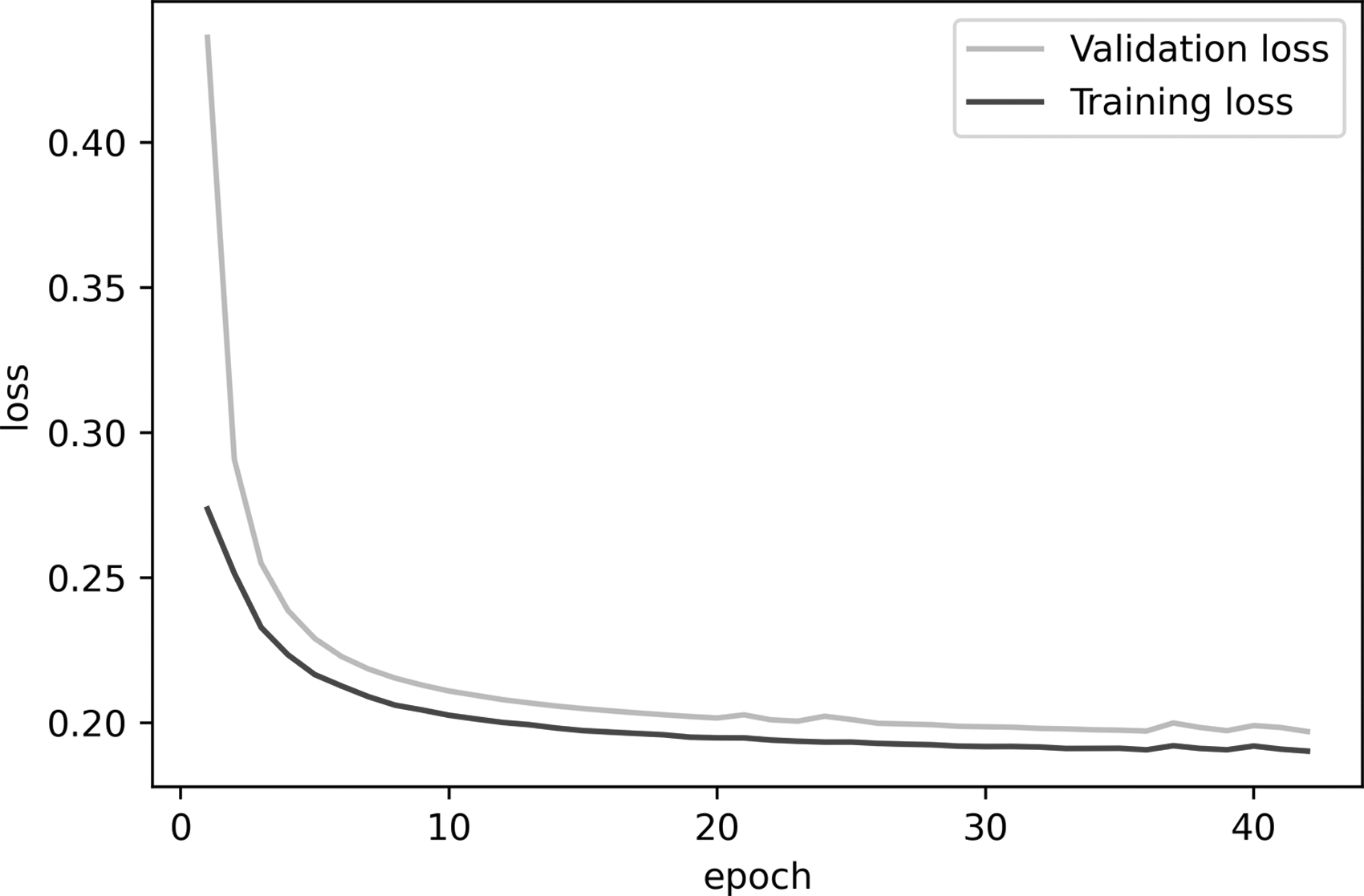
Training/validation loss plot. PC, principal component.
Principal component analysis (PCA) is one of the most widely used techniques to reduce the size of a large dataset by preserving the information in the data. A PCA was performed on the features of the training and the newly generated data to compare these two datasets. First, 24 common physicochemical features were calculated for the original training molecules and the generated virtual molecules. Then, PCA was performed on these generated features, and the first two principal components (PC1, PC2) were selected. The coordinates of the generated molecules were transformed accordingly. Figure 6 shows the original and generated molecules with respect to these principal components. It is observed that the generated molecules are in the same chemical subspace as the original molecules.

Principal component analysis on the training and generated molecules. MW, molecular weight.
MW is obtained by adding the atomic weight of the atoms that make up the molecular formula of the compound. An important molecular feature in drug discovery is molecular hydrophobicity, commonly defined as log P (logarithm of the 1-octanol/water partition coefficient). ClogP is one of the most widely used methods for estimating log P (Ghose et al., 1998). Figure 7 uses Violin-Plot to compare the MW distributions and clogP for the generated and original molecules. Visual inspection shows a close match between the generated and the original molecules. Although the generated molecules are very dense from the original molecules in both diagrams, we see that the medians are similar.
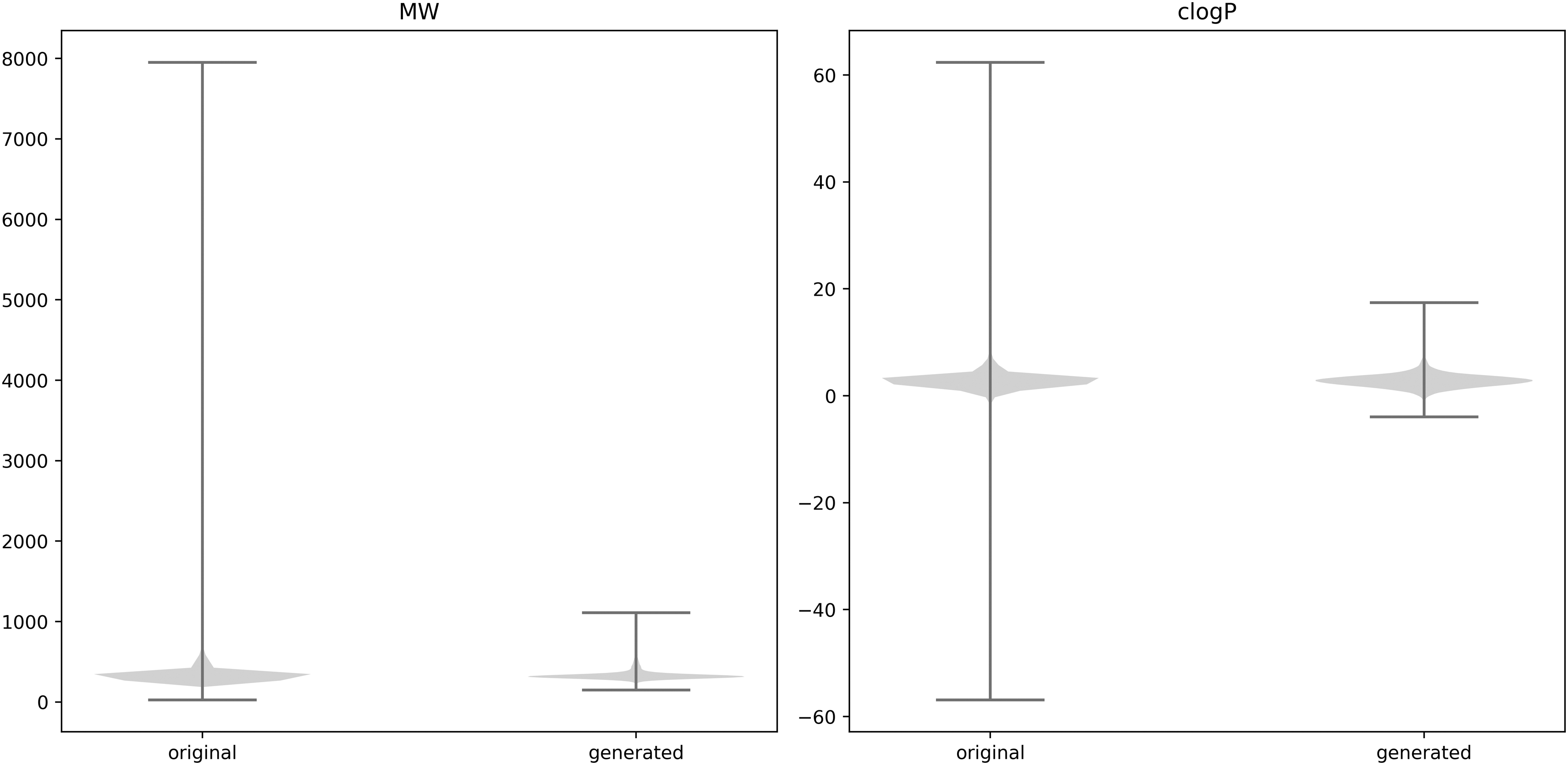
Violin-plots for MW and clogP distributions.
The molecular docking study was performed using Maestro software version 12.5. In this regard, the X-ray crystallographic structure of the SARS-CoV-2 main protease (PDB ID:6LU7) was utilized as an enzyme structure. The novel structures obtained from fine-tuning were docked into the SARS-CoV-2 main protease active site and docking score, values of these structures are presented in a Supplementary Data. Compound AADH with a Dock-score of −9.16 exhibited a lower affinity to the active site in comparison with the N3 inhibitor, potent SARS-CoV-2 main protease inhibitor with a Dock-score of −8.08.
The docking results revealed that the compounds AADH, AADM, AACY, AACQ, AACZ, AAAB, and AACF fit properly in the catalytic pocket with appropriate affinity to the active site (Table 1). The binding interaction of N3 in the active site of protease was evaluated. As shown in Figure 8, the NH group of the N3 on the active site pocket has a suitable distance from the amino acids of Gln189, Glu166, and Thr 190 for effective hydrogen bonding, and additional hydrogen bonds could be formed between the carbonyl moiety and Glu166 and Gly143of the catalytic pocket. In addition, the isopropyl group may have lipophilic binding to Val186 and Leu167 of the active site pocket. As represented in Figure 8, compounds AADH and AADM with higher affinities in comparison with N3 were well accommodated and showed appropriate interactions inside the active site. It was also found that two amino acids of Gln189 and Glu166 have an essential role in inhibitory activities and should be considered in novel drug design. In addition, the α,β-unsaturated carbonyl of the N3 is very close to Cys145, which increases the chances of covalent interaction through the Michael addition's mechanism. It is the potential reason for the high inhibitory activity of this compound and makes this ligand irreversible SARS-CoV-2 main protease inhibitor. Therefore, the inhibitory activity of the mentioned compounds will be improved by adding the group which is able to form covalent addition such as phosphonate, β-lactam, and epoxide groups. The compounds AADM, AACY, AACQ, and AAAB have a common scaffold. Salicylamide moiety might seem to be a suitable precursor for rational anti-COVID-19 drug design. Eventually, the presented seven analogs could be considered as promising hit compounds for corona target and should be further evaluated as a drug-therapeutic candidate.
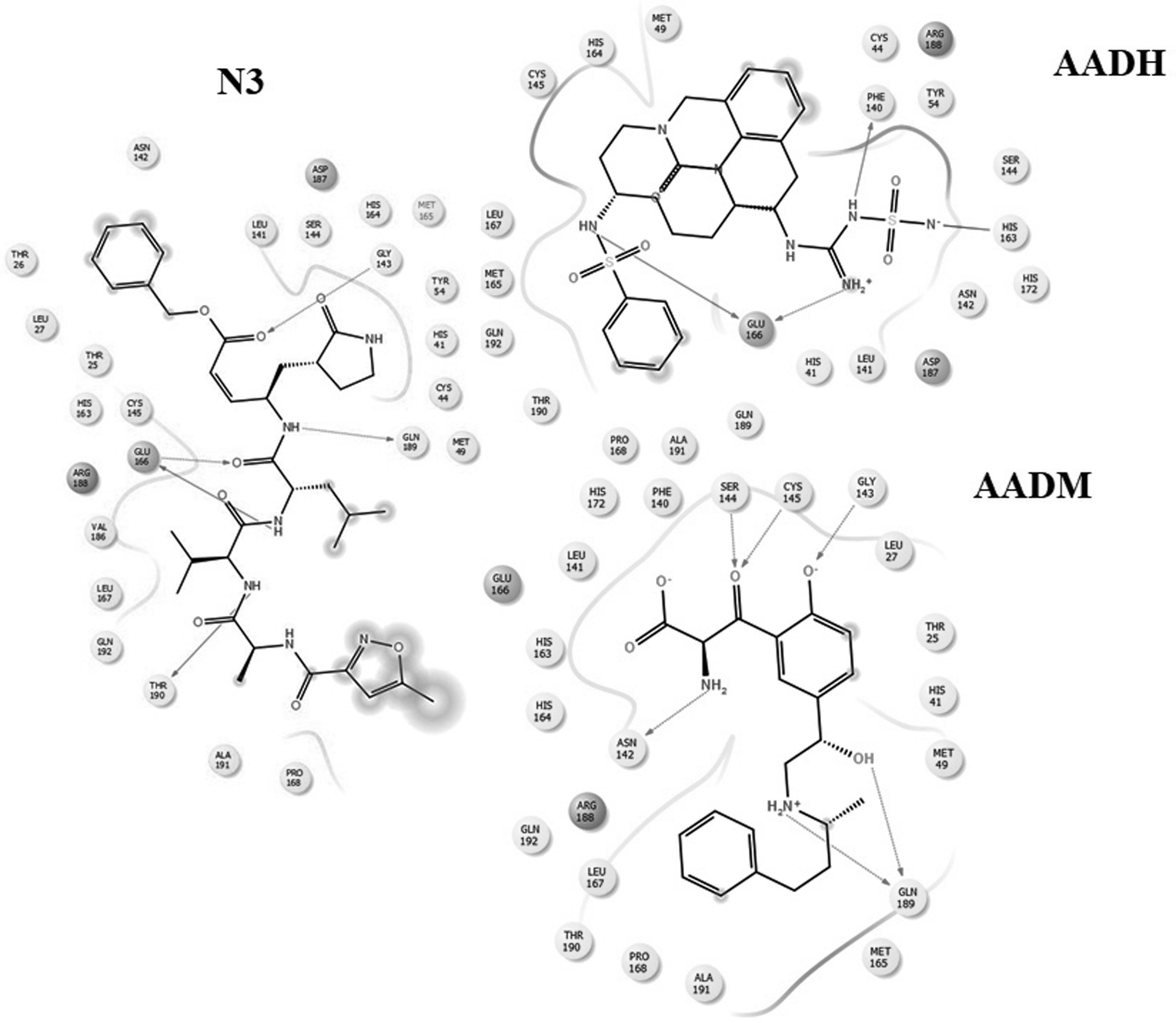
Binding interactions of the inhibitors N3, AADH, and AADM with the active site of the COVID-19 main protease enzyme.
The Results of Docking Studies of the Novel Generated Structures and N3
To evaluate the drug-likeness properties and oral bioavailability of our novel compounds, in-silico ADME studies were applied using the SwissADME online calculator (Table 2). According to Lipinski's rule of five [mLog P (octanol-water partition coefficient) ≤5, MW ≤500, number of hydrogen bond acceptors ≤10, and number of hydrogen bond donors ≤5], an orally active drug candidate has no more than one violation of the criteria (Ertl et al., 2000). Some structures had the potential to be administered through oral route as they abided by Lipinski's rule of five and showed high GI absorption. All compounds have no mutagenic, tumorigenic, and irritant risks; therefore the toxicity of the compounds was acceptable for further investigation.
Predicted Absorption, Distribution, Metabolism, and Excretion Properties of the Novel Generated Structures and N3
Predicted Absorption, Distribution, Metabolism, and Excretion Properties of the Novel Generated Structures and N3
GI, gastrointestinal absorption; mLogP, logarithm of partition coefficient of the compound between n-octanol and water; MW, molecular weight; n-OHNH donors, number of hydrogen bond donors; n-ON acceptors, number of hydrogen bond acceptors; n-ROTB, number of rotatable bonds; TPSA, topological polar surface area.
In this section, 120 ns MD trajectories of the eight main protease complexes, namely, AADH, AADM, AACY, AACQ, AACZ, AAAB, AACF, and N3 as the control model, are analyzed, Figure 9. To evaluate the reliability of our MD simulations for the considered complexes, the RMSD of the enzyme's backbone heavy atoms over their 100-ns MD trajectories is analyzed (Fig. 9A). As the Figure reveals, all the systems are stabilized and achieved convergence over 120 ns of simulations run. This analysis suggests that the generated trajectories of all models are reliable. The average values of the RMSD for the ligand-bound complexes of the main protease with AADH, AADM, AACY, AACQ, AACZ, AAAB, AACF, and N3 are 1.93 Å, 2.52 Å, 2.51 Å, 1.70 Å, 1.85 Å, 2.01 Å, 2.37 Å, and 2.05 Å, respectively. As it is observed in Figure 9A, the RMSD plot shows reasonable convergence particularly after 20 ns, which indicates the stability of the systems during the MD trajectories.
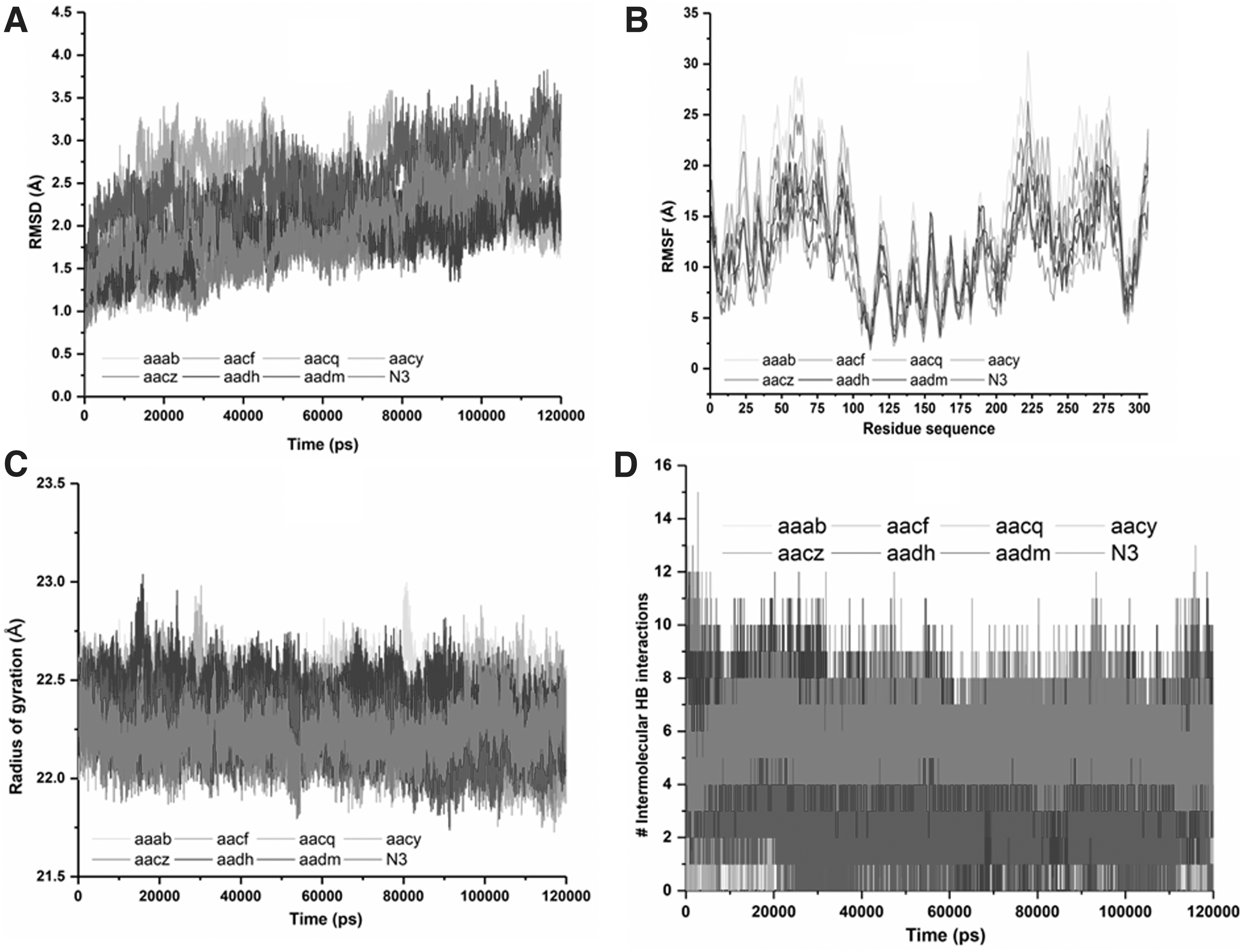
To provide detailed insight into the structural fluctuation and flexibility of different regions of the amino acid residues of the main protease enzyme upon binding of the selected compounds RMSF values for Cα atoms are calculated from trajectories generated over 120 ns of MD trajectories (Fig. 9B). As it is depicted, AADH, AADM, AACY, AACQ, AACZ, AAAB, AACF, and N3 exhibit the average fluctuations in the residues with 12.86 Å, 12.84 Å, 11.73 Å, 10.37 Å, 9.85 Å, 15.54 Å, 13.52 Å, and 13.56 Å, respectively (Fig. 9B). According to the RMSF plot, it is evident that the complexes of AACQ, AACZ, and AACY imposed less overall structural flexibility and residual fluctuations upon binding which confirms less structural flexibility upon binding, while AADH, AADM, AAAB, AACF, and N3 induced higher residual fluctuations upon binding. The variations in fluctuations might have an impact on the total binding affinities upon complexation.
The Rg parameter determines the structural compactness of the system through the overall conformational variations in the complex structure in terms of the distance from the center of mass of the complex. The mediocre values of Rg for AADH, AADM, AACY, AACQ, AACZ, AAAB, AACF, and N3 complexes are noted to be 2.39 Å, 22.21 Å, 22.32 Å, 22.36 Å, 22.27 Å, 22.37 Å, 22.19 Å, and 22.22 Å, respectively (Fig. 9C). The mean Rg values do not vary significantly for the selected compounds in complex with the main protease enzyme (Fig. 9C). This collectively reflects rather moderate conformational changes for the inhibitor-enzyme complexes during the simulations and suggests that the presence of the bound inhibitor dampens the dynamic motion of the macromolecule. This observation is certainly expected based on the number of stabilizing noncovalent interactions between the enzyme and each inhibitor.
To confirm the overall stability and strength of the interactions of the enzyme-ligand complex, we monitored the intermolecular hydrogen bonds between the enzyme and the bound inhibitor (Fig. 9D). Intermolecular hydrogen bonding between the protein and the ligand plays a vital role in stabilizing all protein-ligand complexes. The stability of the hydrogen bond network formed between the investigated ligands and the enzyme was evaluated through the 120-ns MD simulation trajectory. Detailed time-dependent information on the hydrogen bonding pattern, specifically the number of intact hydrogen bonds, between the protein and bound ligand is presented in Figure 9D. The average number of intermolecular hydrogen bonds formed between the ligand and the enzyme within the binding pocket of the main protease is observed to be 5, 5, and 6 in complexes AADH, AACF, and N3 over the complete 120 ns MD trajectory (Fig. 9D). However, in the case of AADM, AACY, AACQ, AACZ, and AAAB the average number of intermolecular hydrogen bonding interactions was 3, 2, 3, 2, and 3, respectively. These observations likely explain the tighter binding interactions and, overall, more stable complexes in terms of binding affinities in complexes with a higher number of hydrogen bonding interactions.
To understand the impact of inhibitors upon complexation in terms of their binding affinities, MM-GBSA binding free energy method was utilized to calculate the binding free energies and their components of the complexes, Table 3. As it is evident in Table 3, the total binding free energies (ΔGbind) of AADH, AADM, AACY, AACQ, AACZ, AAAB, AACF, and N3 are −67.06 kcal/mol, −20.73 kcal/mol, −23.99 kcal/mol, −21.96 kcal/mol, −18.81 kcal/mol, −39.83 kcal/mol, −47.07 kcal/mol, and −51.91, respectively. Accordingly, among all the studied complexes, AADH, N3, and AACF depicted the lowest values of ΔGbind. At this point, it is interesting to address the key contributions that each binding component can impose on the total binding free energies. It is evident that among the studied complexes, the ΔGgas as the favorable contributing index to the total ΔGbind has the lowest values for AADH (−115.47 kcal/mol), N3 (−98.93 kcal/mol), and AACF (−79.46 kcal/mol) complexes. This observation implies the most favorable contribution values of ΔEvdw for AADH (−56.79 kcal/mol), N3 (−56.60 kcal/mol), and AACF (−39.32) into the total binding free energies. Interestingly, this observation revealed another key contributing component contribution of ΔGnonpolar for the complexes of AADH (−7.33 kcal/mol) and N3 (−7.50 kcal/mol), and AACF (−5.59) which leads to lowest ΔGbind values for these complexes. According to the obtained energy component results, it could be inferred that the ΔEvdw and ΔGnonpolar have the dominant contribution into the binding affinities for the studied complexes.
Binding Free Energies and Their Components for the Studied Complexes Using the Molecular Mechanics/Generalized Born Surface Area Method
The energy components are in kcal mol−1.
To understand the impact of inhibitors upon complexation in terms of their binding affinities, MM-GBSA binding free energy method was utilized to calculate the binding free energies and their components of the complexes, Table 3. As it is evident in Table 3, the total binding free energies (ΔGbind) of AADH, AADM, AACY, AACQ, AACZ, AAAB, AACF, and N3 are −67.06 kcal/mol, −20.73 kcal/mol, −23.99 kcal/mol, −21.96 kcal/mol, −18.81 kcal/mol, −39.83 kcal/mol, −47.07 kcal/mol, and −51.91, respectively. Accordingly, among all the studied complexes, AADH, N3, and AACF depicted the lowest values of ΔGbind. At this point, it is interesting to address the key contributions that each binding component can impose on the total binding free energies. It is evident that among the studied complexes, the ΔGgas as the favorable contributing index into the total ΔGbind has the lowest values for AADH (−115.47 kcal/mol), N3 (−98.93 kcal/mol), and AACF (−79.46 kcal/mol) complexes. This observation implies the most favorable contribution values of ΔEvdw for AADH (−56.79 kcal/mol) and N3 (−56.60 kcal/mol), and AACF (−39.32) into the total binding free energies. Interestingly, this observation revealed another key contributing component contribution of ΔGnonpolar for the complexes of AADH (−7.33 kcal/mol) and N3 (−7.50 kcal/mol), and AACF (−5.59) which leads to the lowest ΔGbind values for these complexes. According to the obtained energy component results, it could be inferred that the ΔEvdw and ΔGnonpolar have the dominant contribution to the binding affinities for the studied complexes.
CONCLUSION
In recent years, RNN based deep learning method is frequently used in drug design and virtual screening. In this study, we presented a model to generate drug candidates using the LSTM network. After training and fine-tuning the proposed model, we used this model for generating compounds that can act as SARS-CoV-2 inhibitors. The results of investigations showed that the generative LSTM network could learn the SMILES grammars of the known SARS-CoV-2 inhibitors and generate the novel potential inhibitors, efficiently. Furthermore, the seven best compounds in the docking study were considered for in silico calculation of drug-likeness properties. ADME studies revealed that compounds AADM and AACY with significant binding affinities to SARS-CoV-2 main protease have appropriate physicochemical properties and might be administrated as orally active agents. MD simulations approach revealed that the two highly selective compounds, namely, AADH and AACF possessed significant binding affinity and presumably inhibition of the target. Based on our overall observations, compounds AADH and AACF could be recommended as potential leads against the SARS-CoV-2 main protease enzyme. Based on the obtained finding, the presented seven analogs might be a worthy candidate for future evaluations to develop novel potent COVID inhibitors.
Footnotes
ACKNOWLEDGMENTS
Z.F. thanks Prof. Orde Q. Munro at Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand (Johannesburg-South Africa), for providing CHPC access under his principal investigator role and the CHPC server based in Cape Town (South Africa) for computational resources.
AUTHORs' CONTRIBUTIONS
A.M.: Conceptualization, software, and writing-initial draft. E.R.: Writing—review and editing, result analysis, and supervision. S.G. and Z.F.: MD simulation. S.M.M.: Writing—review and editing, result analysis, and supervision.
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
This work was supported by the research council of the school of pharmacy, Shahid Beheshti University of medical sciences (Grant No. 43002969).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.