
Abstract
Drug–drug interaction (DDI) is a key concern in drug development and pharmacovigilance. It is important to improve DDI predictions by integrating multisource data from various pharmaceutical companies. Unfortunately, the data privacy and financial interest issues seriously influence the interinstitutional collaborations for DDI predictions. We propose multiparty computation DDI (MPCDDI), a secure MPC-based deep learning framework for DDI predictions. MPCDDI leverages the secret sharing technologies to incorporate the drug-related feature data from multiple institutions and develops a deep learning model for DDI predictions. In MPCDDI, all data transmission and deep learning operations are integrated into secure MPC frameworks to enable high-quality collaboration among pharmaceutical institutions without divulging private drug-related information. The results suggest that MPCDDI is superior to other eight baselines and achieves the similar performance to that of the corresponding plaintext collaborations. More interestingly, MPCDDI significantly outperforms methods that use private data from the single institution. In summary, MPCDDI is an effective framework for promoting collaborative and privacy-preserving drug discovery.
INTRODUCTION
Drug–drug interaction (DDI) refers to that when taking multiple drugs at the same time, the activity and effectiveness of drugs are affected by other drugs (Bijnsdorp et al., 2011). Drug interactions may lead to adverse drug reactions (ADRs), which may lead to unexpected effects, resulting in massive morbidity and mortality (Vilar et al., 2014a). It is important to effectively recognize potential DDIs that can maximize synergistic benefits and reduce ADRs (Edwards and Aronson, 2000b) when treating a disease. Since testing DDI in a wet laboratory is expensive and time-consuming, it is urgent to develop computational methods for accurately predicting potential DDIs.
To accurately predict potential DDIs, many machine learning and deep learning methods have been proposed. In particular, various machine learning models have achieved excellent performance for DDI predictions. Cheng and Zhao (2014) calculated the similarity of drug pairs by phenotypic similarity, therapeutic similarity, chemical structural similarity, and genomic similarity, and then five prediction models are applied: naive Bayes, decision tree, k-nearest neighbor, logistic regression, and support vector machine to predict DDI. Similarly, Song et al. (2019) developed a support vector machine approach based on a set of similarity measures, including 2D molecular structure similarity, 3D pharmacodynamic similarity, interaction fingerprint (IPF) similarity, target similarity, and adverse drug effect (ADE) similarity.
Mei and Zhang (2021) proposed a simplified drug target profile representation to describe drugs and drug pairs. They subsequently utilized a l2-regularized logistic regression model to predict drug–drug interactions. With the rapid development of AI, deep learning has achieved great success in various fields and has been widely used in DDI prediction (Min et al., 2016). Ma et al. (2018) proposed an attentive multiview graph autoencoders-based drug similarity integration for DDI predictions in which the weights of each view are determined by an attentive mechanism. Zitnik et al. (2018) proposed Decagon for predicting ADRs, which creates a brand-new multimodal graph convolutional neural network. Deac et al. (2019) predicted drug–drug adverse effects by utilizing a co-attentional mechanism, which was able to get advanced results.
Nyamabo et al. (2022) used the gated message-passing neural network (GMPNN) to learn the chemical substructures of drugs from the molecular graph of drugs. DDI prediction is based on the interactions between pairs of their substructures (Nyamabo et al., 2022). Nyamabo et al. (2021) proposed a deep learning framework called substructure–substructure interaction–DDI (SSI–DDI), which directly learns the original molecular diagram representation of drugs and predicts DDI based on their respective substructures. Wang et al. (2021) proposed a multiview graph contrastive representation learning to capture inter-view molecule structure and intra-view interactions between drug molecules. Each node in the interaction graph itself is a drug molecular graph, which can be learned as drug properties by GCN (Wang et al., 2021).
Feng et al. (2020) proposed a robust method DPDDI to predict potential DDIs, which only learns DDI network information through GCN without considering any drug properties. Pang et al. (2022) proposed a novel attention-mechanism-based multidimensional feature encoder to learn the representation of drug features from multiple dimensions, including smiles sequence and drug molecular graph information. The effectiveness of deep learning methods for predicting DDIs has been widely recognized due to their impressive predictive capabilities. However, it is important to note that many previous models have heavily relied on large-scale drug data. Unfortunately, obtaining high-quality drug data is a challenging and expensive task in the field of drug discovery.
As a result, the practical application of these deep learning-based DDI prediction models in drug development scenarios may not meet the desired standards (Wang et al., 2022). With the accumulation of biomedical data, there are various publicly available bioinformatics and pharmacogenomics databases. For instance, DrugBank (Law et al., 2014) is a comprehensive database for drug discovery, which plays a significant role in drug discovery by offering comprehensive information on drugs and their respective targets. PDBbind database serves as a comprehensive repository of binding affinity data derived from experimental measurements of protein-ligand complexes housed within the protein database (Liu et al., 2015).
Drug Repurposing Knowledge Graph (DRKG) (Ioannidis et al., 2020) is a comprehensive biological knowledge graph, which provides abundant relational data among genes, compounds, diseases, biological processes, side effects, and symptoms. KEGG (Kanehisa et al., 2010) is a large-scale molecular data set used to understand the advanced functions of biological systems. The integration of diverse data sources from both public databases and private entities, such as pharmaceutical companies and academic institutions, has been identified as a promising approach to enhance the accuracy and reliability of DDI predictions. However, concerns regarding drug data privacy have hindered the disclosure of valuable and proprietary data by pharmaceutical companies.
To address the challenges associated with data sharing, researchers have made strides in implementing secure multiparty computation (MPC) techniques (Cho et al., 2018; Hie et al., 2018; Jagadeesh et al., 2017; Ma et al., 2020). These approaches aim to foster data sharing and collaboration within domains such as drug discovery and other biomedical fields. By leveraging MPC techniques, privacy-preserving computations can be carried out on distributed data without the need for direct data exchange. This allows multiple parties to collectively analyze and extract insights from their respective data sets while preserving the confidentiality of sensitive information. The application of MPC techniques in the biomedical domain holds promise for advancing research and facilitating collaborations across different organizations, including pharmaceutical companies, academic institutions, and research consortia.
Cho et al. (2018) leveraged MPC techniques to conduct large-scale genome-wide analysis while safeguarding the privacy of sensitive data, including genotypic and phenotypic information. Hie et al. (2018) introduced a predictive model, which leverages MPC techniques to ensure the privacy and confidentiality of sensitive data involved in the training process. Unfortunately, the application of MPC protocol-based deep learning techniques in the field of drug discovery remains somewhat constrained. Specifically, the development of MPC-based deep learning methods for accurate predictions of DDI poses a considerable challenge.
In this study, we propose a secure MPC-based deep learning framework MPCDDI for DDI predictions. MPCDDI facilitates the integration of drug-related data among multiple pharmaceutical companies and institutions through the utilization of secret sharing technology. This ensures the confidentiality and security of the shared information. To enhance collaboration and privacy in drug discovery, MPCDDI incorporates a deep neural network that consists of a projection model and multilayer perceptrons (MLPs) for predicting DDIs. Importantly, all deep learning operations within MPCDDI are executed under MPC frameworks.
This approach not only promotes collaboration among pharmaceutical institutions but also protects the privacy of sensitive data during the drug discovery process. The experimental results demonstrate that MPCDDI outperforms eight existing methods and achieves comparable results with those obtained through plaintext collaborations. These findings underscore the effectiveness of the MPCDDI framework in facilitating collaborative and privacy-preserving drug discovery efforts. In conclusion, the MPCDDI framework serves as a valuable tool for fostering collaborative endeavors in the field of drug discovery while ensuring the protection of sensitive information.
METHODS
The schematic workflow of MPCDDI, as illustrated in Figure 1, provides a visual representation of its application scenario. In this particular scenario, four distinct pharmaceutical institutions possess diverse sets of feature data pertaining to drugs. The primary objective of the MPCDDI framework is to seamlessly integrate these disparate feature data sources, enabling the four pharmaceutical institutions to collectively enhance DDI predictions. Notably, this integration is accomplished without compromising the confidentiality of any sensitive or confidential data.
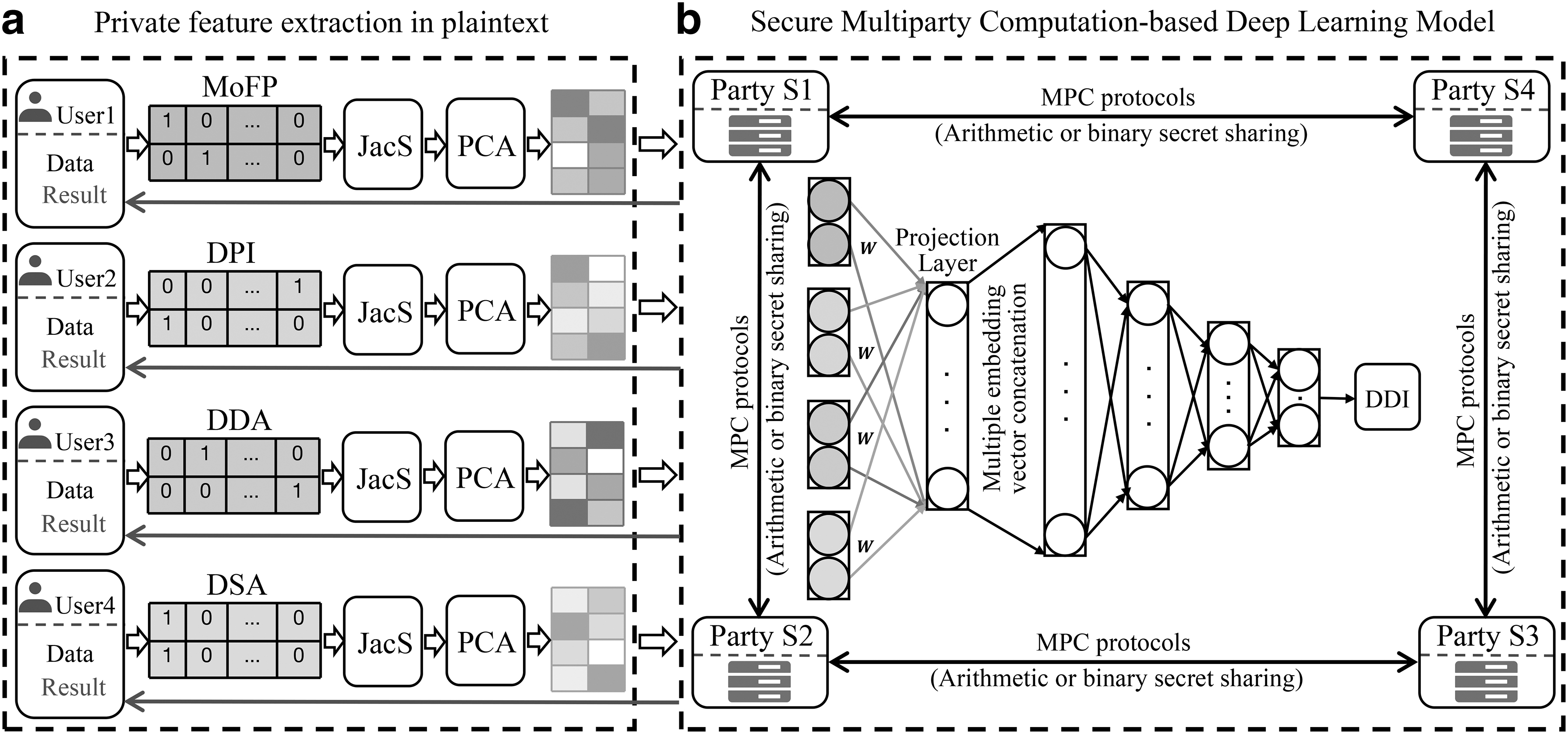
The schematic workflow of MPCDDI. MoFP, DPI, DDA, and DSA denote the MoFPs, drug–protein interactions, DDAs, and DSAs, respectively. JacS and PCA represent the Jaccard similarity and principal component analysis, respectively.
In this study, secure MPC is treated as a user-server model, where each pharmaceutical institution with private data is represented by a also named user. There are four users (e.g., pharmaceutical institutions) that is represented by
Based on the aforementioned assumptions, we carry out MPC by two key steps. In the first stage, the private data of each user is divided into four parts and then is sent to four servers
In the second stage, we break down the deep learning algorithm into a set of fundamental operations including private addition, private multiplication, comparison, and a few linear and nonlinear functions. Each party fails to capture the information about input data when executing a basic private operation. The four parties carry out a series of private actions sequentially to finish the assignments. To increase efficiency, some private activities can be run concurrently. The confidentiality of input is preserved because neither side can discover the input, intermediate values, or final results from either secret shares or private operations.
All the basic private operations are implemented by using the protocols available in CRYPTEN that adopts binary secret sharing (Goldreich et al., 2019), arithmetic secret sharing (Damgård et al., 2012), and conversions between these two types of sharing to facilitate secure computations. Most of basic operations, such as bilinear operations and multiplications, adopt arithmetic secret sharing mechanisms. Similarly, binary secret sharing techniques are used for the evaluation of logical expressions, such as the activation function rectified linear units (ReLU).
Some other operations involve the conversion between arithmetic secret sharing and binary secret sharing, which can be achieved by the ABY share-conversion techniques (Demmler et al., 2015). At the cryptographic level, Beaver multiplication triples (Beaver, 1991) can help implement basic arithmetic operations, whereas the GMW protocol (Goldreich et al., 2019) proposed by Goldreich, Micali, and Wigderson can help implement binary circuit evaluation. The four parties acquire the secret results after finishing all the private activities. Finally,
Private feature extraction
In this study, we assume that there are four pharmaceutical institutions that, respectively, have different drug data, that is, molecular fingerprints (MoFPs) which are derived by RDKit (Landrum, 2013), drug–side effect associations (DSAs), drug–disease associations (DDAs), and drug–protein interactions. Four binary feature vectors can be used to represent a given drug. The values (1 or 0) in feature vectors denote the presence or absence of the relevant descriptors. Based on these different drug descriptors, four kinds of drug–drug similarity are computed by the Jaccard similarity metric and treated as four kinds of drug features, respectively.
In real drug discovery, there are a great number of drugs or molecules, thus leading to high-dimension and sparse drug similarity features. To overcome this problem, all similarity features are fed into principal component analysis for reducing the dimensionality of each drug. The low-dimensional features of drugs in each user are secretly shared with various parties
MPC-based deep learning
Each party obtain four secret features
where
where W is the weight matrix of linear transformations, b is a bias vector, and
The DDI prediction module adopts an MLP with three fully connected layers to predict DDIs. The two hidden layers and output layer employ ReLU and sigmoid activation function, respectively. The batch normalization is used to increase the speed of convergence. Furthermore, we add the dropout layers (Srivastava et al., 2014) to improve generalization performance and reduce overfitting. By fully utilizing the drug feature data from all users to forecast DDIs, MPCDDI trains the deep learning models using private operation protocols, while only disclosing the predicted actions to the corresponding users. After running MPCDDI, each user will obtain the predicted DDIs and trained deep learning models.
The proposed MPCDDI is implemented based on CRYPTEN framework (Knott et al., 2021) to realize DDI predictions from private drug data under MPC protocol. The CRYPTEN framework provides a secure computational backend for PyTorch, while still retaining the PyTorch front-end APIs that enable experimentation with deep neural networks. The framework is similar to PyTorch while all values are secretly shared among the modes of multiple parties.
Data sets
In this study, we used a heterogeneous data set (Luo et al., 2017) in which include the DDIs and the drug–protein interactions network derived from DrugBank 3.0 (Knox et al., 2010). The DDA network was derived from the Comparative Toxicogenomics Database (Davis et al., 2013). The DSAs were obtained from the Comparative Toxicogenomics Database and SIDER (Kuhn et al., 2010). Moreover, the simplified molecular input line entry system of each drug was converted into a 1024-bit vector, known as the MoFPs, using the RDKit program (Landrum, 2013). The resulting MoFP contained information about the drug structure. The sizes of the heterogeneous data set are presented in Table 1. In MPCDDI, we consider MoFP, DPI, DDA, and DSA as the private feature of four users.
The Sizes of Heterogeneous Data Set
The Sizes of Heterogeneous Data Set
To evaluate the performance of MPCDDI, it was compared with eight methods, including DDIMDL (Deng et al., 2020), DeepDDI (Ryu et al., 2018), LINE (Tang et al., 2015), GraRep (Cao et al., 2015), struc2vec (Ribeiro et al., 2017), GMPNN-CS (Nyamabo et al., 2022), SSI-DDI (Nyamabo et al., 2021), and MIRACLE (Wang et al., 2021) models. DDIMDL, DeepDDI, GMPNN-CS, SSI-DDI, and MIRACLE are deep learning-based DDI prediction methods and have achieved greater performance on various data sets. LINE, GraRep, and struc2vec, as classic graph embedding methods, have also been extensively applied in the biomedical field. All identified DDIs in the data set were considered as positive samples. To obtain negative samples, a random selection method was employed for the unidentified interactions.
It should be noted that the number of negative samples is consistent with the number of positive samples, and these samples were then divided into two sets: a testing set comprising 10% of the samples, and a training set comprising the remaining 90%. In this study, the task of DDI prediction was approached as a binary classification problem. Consequently, the evaluation of the predictive models was based on several performance metrics, including the area under the receiver operating characteristic curve (AUROC), recall, and the precision–recall curve (AUPR). These metrics were considered suitable for assessing the effectiveness and discriminatory power of the predictive models.
In this study, the representation vectors generated by LINE, GraRep, and struc2vec were fed into Logistic Regression models for DDI predictions. For hyperparameters of MPCDDI, we set 256 neurons in the projection layer. The MLP includes three hidden layers for DDI predictions, where there are 128, 64, and 2 neurons in each layer, respectively. We used Adam optimizer with a 1e-4 learning rate, and set the dropout rate at 0.3. The hyperparameters of the LINE, GraRep, and struc2vec models were set as the default values in Yue et al. (2020), because Yue et al. carefully optimized them by grid search. The parameters of DDIMDL, DeepDDI, GMPNN-CS, SSI-DDI, and MIRACLE were selected as default based on their respective articles (Deng et al., 2020; Nyamabo et al., 2022; Nyamabo et al., 2021; Ryu et al., 2018; Wang et al., 2021).
DDIMDL: It employs a joint mechanism to effectively amalgamate a diverse range of drug features for accurate DDI predictions. Within this framework, the number of units in the current layer was established as half the number present in the previous layer. Furthermore, the batch size was set to 256, whereas the dropout rate was set to 0.3.
DeepDDI: It first involves computing structure similarity profiles for drugs using MoFPs. These profiles capture the structural characteristics of the drugs and are then fed into deep neural networks for predicting DDI. The DeepDDI architecture consists of nine hidden layers, with each layer containing 1024 nodes. During model training, a batch size of 256 and a learning rate of 0.0001 were utilized.
LINE: It employs a joint training approach to capture both local and global network structures by optimizing first-order similarity and second-order similarity objective functions. This versatile algorithm can be applied to various information networks. In LINE, the negative ratio was set to 5, and the order parameter was set to 2.
GraRep: It introduces an improved loss function that effectively captures intricate local and global structural characteristics within the biomedical network. Specifically, GraRep employs a fixed value of k-step, set to 4, and a learning rate of 0.01.
struc2vec: It defines node similarity from the perspective of spatial structural similarity. In struc2vec, the length of the random walk was set to 64, the window size was set to 10, and the number of random walks was set to 32.
GMPNN-CS: It is a gated message passing neural network for learning various structures from the drug molecular diagram, which is then fed into the downstream model to predict the DDI between a pair of drugs. In GMPNN-CS, we set the batch size as 512, learning rate as 0.001, dropout as 0, and the number of message passing steps as 3.
SSI-DDI: The method first uses rdkit (https://www.rdkit.org) tool to obtain drug molecular graph through drug smiles sequence, then uses graph neural network to learn the original molecular diagram representation of drugs, and finally predicts DDI based on their respective substructure characteristics. Since the original method is used for the multiclassification task of DDIs, we changed the multiple drug interaction types in the code to only one interaction type. For other parameters, learning rate is set to 0.01, batch size is set to 32, and the number of hidden features is set to 64.
MIRACLE: It regards the DDI network as a multiview graph, in which each node itself in the interaction graph is a drug molecular graph. MIRACLE uses GCN to learn DDI diagrams and bond-aware aggressive message propagating method to learn drug molecular diagrams. In MIRACLE, the dimension of hidden layers is set to 300, the number of message-passing steps is set to 3, and the number of nodes in three hidden layers of gcn is set to 100, 81, and 64, respectively.
Comparisons between MPCDDI and baselines
The results of MPCDDI and baselines are shown in Figure 2. We found that MPCDDI (AUROC = 0.938, AUPR = 0.942, recall = 0.870) was superior to baselines on all evaluation metrics, with 4.65% higher AUROC, 6.54% higher AUPR, and 3.49% higher recall than the average performance of the eight baselines. In particular, MPCDDI outperformed GMPNN-CS with 5.5%, 8.6%, and 12.1% improvements in terms of recall, AUROC, and AUPR for DDI predictions, respectively. These results suggested that MPCDDI can achieve better performance than other methods.
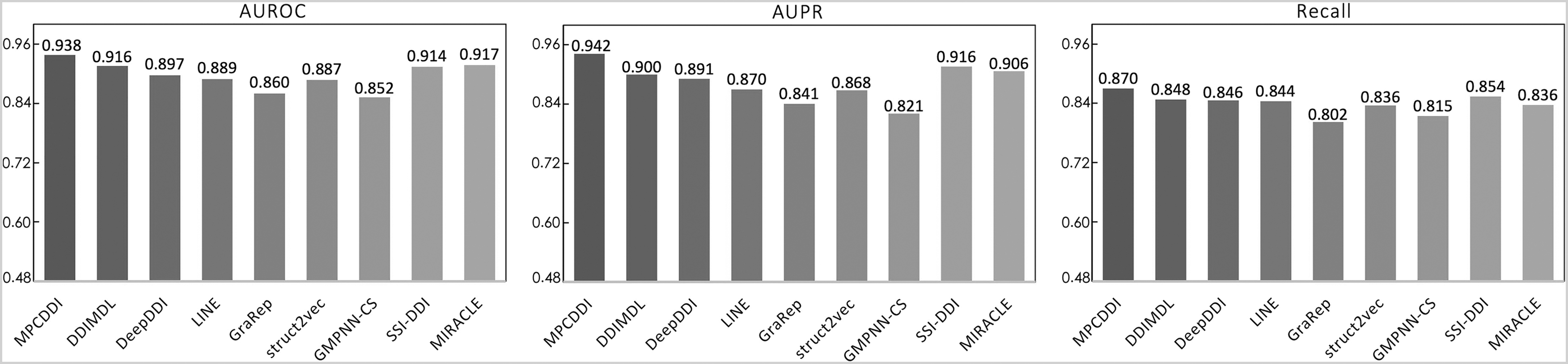
The results of MPCDDI and baseline methods for DDI predictions. AUPR, area under the precision–recall curve; AUROC, area under the receiver operating characteristic curve; GMPNN, gated message-passing neural network; MPCDDI, multiparty computation drug–drug interaction; SSI, substructure–substructure interaction.
Our research investigates whether our MPC collaboration protocol results in a reduction in prediction accuracy compared with the corresponding plaintext learning method (named PlainDDI) in the context of open collaboration utilizing all shared information. To be specific, PlainDDI used four plaintext features to drive the proposed deep learning model for DDI predictions. The performance comparison between MPCDI and PlainDDI is presented in Table 2, with the best results highlighted in bold. We observed that there was no significant difference in performance between MPCDI and PlainDDI.
The Performances of MPCDDI and PlainDDI
The Performances of MPCDDI and PlainDDI
Best results highlighted in bold.
AUPR, area under the precision–recall curve; AUROC, area under the receiver operating characteristic curve;
For example, the disparity between MPCDI and PlainDDI was only 0.6% in terms of AUROC, whereas MPCDI exhibited a 2.3% advantage over PlainDDI in terms of AUPR. These results suggested that MPCDI facilitates high-quality collaboration within pharmaceutical organizations, while maintaining comparable performance with plaintext prediction models on publicly shared information. In other words, MPCDI not only delivers superior results but also ensures the privacy of sensitive data within pharmaceutical organizations.
In a perfect collaborative situation, medication feature data from several pharmaceutical institutions are combined to create a multimodal feature space that is conducive to training better deep learning models. In MPCDDI, we imitated the application scenario where four different data (i.e., MoFP, DPI, DDA, and DSA) were randomly distributed to four pharmaceutical companies and treated as their private feature data. These private features from different institutions were integrated through MPC protocols and then used to jointly training a deep learning model for DDI predictions. Therefore, MPCDDI was converted to Fing-DDI without MPC protocols, that is, only using the MoFPs data owned by a single institution. Similarly, MPCDDI was transformed to Pro-DDI, Dis-DDI, and Se-DDI. In Table 3, we described the results of MPCDDI and other methods using only a single feature (i.e., MoFP, DPI, DDA, and DSA).
The Results of MPCDDI and Other Methods Using Private Data Owned by a Single Institution
The Results of MPCDDI and Other Methods Using Private Data Owned by a Single Institution
Best results are marked in boldface.
The best results are marked in boldface. We find that Fing-DDI achieved relatively high results than other three methods (i.e., Pro-DDI, Dis-DDI, and Se-DDI). For example, Fing-DDI obtained 8.4% higher AUROC, 6.8% higher AUPR, and 16.3% higher recall than Pro-DDI for DDI predictions. Furthermore, we observed that MPCDDI consistently achieved higher performance than other methods. Interestingly, MPCDDI generates 5.5%–13.7% AUROC, 7.7%–14.5% AUPR, and 3.2%–20.6% recall improvements than other methods for DDI predictions. In particular, MPCDDI is superior to Pro-DDI, with AUROC, AUPR, and recall improvements of 13.7%, 14.5%, and 20.6%, respectively. These results suggested that MPCDDI can help pharmaceutical institutions or organizations to achieve better prediction performance without divulging private information.
In this part, we assessed the impact of several pharmacological properties on DDI predictions under the encrypted state. MPCDDI is converted to FPD-MPCDDI when we use only three features, including MoFP, DPI, and DDA. Similarly, MPCDDI is converted to PDS-MPCDDI, FDS-MPCDDI, and FPS-MPCDDI. The results of MPCDDI and different feature models are shown in Figure 3. We observe that PDS-MPCDDI obtained relatively poorer results than other three methods (i.e., FDS-MPCDDI, FPS-MPCDDI, and FPD-MPCDDI).

The performance of MPCDDI with different feature combinations. FDS, use of MoFP, DDA, and DSA as the features; FPD, use of MoFP(F), DPI(P), and DDA(D) in MPCDDI; FPS, use of MoFP, DPI, and DSA as the features; PDS, use of DPI, DDA, and DSA as the features.
In particular, FDS-MPCDDI was 2.9% higher AUROC, 3.4% higher AUPR, and 5.3% higher recall than PDS-MPCDDI for DDI predictions. However, we found that FDS-MPCDDI (AUROC = 0.928, AUPR = 0.934, and recall = 0.863) obtained the most similar results to MPCDDI. Furthermore, we observed that MPCDDI achieved higher performance than other four methods. These results suggested that the combinations of four features are contribute to DDI predictions, and that MoFPs have the greater impact on DDI predictions relative to the drug–protein interactions.
To the best of our knowledge, most of the existing DDI prediction methods can be divided into similarity-based methods, graph-based methods, and data integration-based methods. DeepDDI (Ryu et al., 2018) use the SMILES sequence of drugs to generate SSP, and then the SSP of two combined drugs is reduced to and merged into a single vector, which is input into the downstream deep neural network (DNN) for DDI prediction. Vilar et al. (2013) proposed DDIs prediction method based on drug interaction profile fingerprints, which were generated based on DrugBank database. One year later, Vilar et al. (2014b) proposed another similarity-based method that calculates drug similarity from multiple sources, including 2D and 3D molecular structure, interaction profile, target, and side-effect similarities.
Graph-based methods can be further divided into DDI network-based methods, drug molecular graph-based methods, and biological knowledge graph-based methods. DDI network can be easily interpreted as a graph, where the nodes are drugs and the edges are drug interactions. Feng et al. (2020) proposed a DDI network-based method, which learns the topological features of drugs in the DDI network by GCN, and then concatenates the topological features of any two drugs as the feature vectors of a DDI, which is fed into the downstream DNN network to predict DDI. Drug molecules can also be described as graphs, where the nodes are atoms and the edges are bonds between atoms.
Wang et al. (2021) regard drug molecules as molecular graphs and use bond-aware attentive message propagating method to capture drug molecular structure information, which is regarded as the node feature in DDI network. Pang et al. (2022) encode drug features from drug SMILES through two channels, including using frequent consecutive subsequences to encode SMILES sequence and message passing attention network to encode drug molecular graph. Nyamabo et al. (2021) directly learn the original molecular graph representation of drugs and predict DDI based on their respective representations.
Nyamabo et al. (2022) also used the GMPNN to learn the substructures from the molecular graph of drugs. DDI prediction is based on the substructures from pair of drugs (Nyamabo et al., 2022). A biological knowledge graph provides abundant relational data among drugs, genes, diseases, side effects, and so on. Yu et al. (2021) proposed a method SumGNN, which maps drug pairs to a subgraph of potential biomedical entities in KG and learns the subgraph by GNN to predict DDIs. Hao et al. (2023) proposed a method 3WDDI, which combines knowledge map embedding as a supplementary feature to enhance DDI prediction.
Many methods improve the performance of DDI prediction by data integration. Cheng and Zhao (2014) calculated the similarity of drug pairs by five types of similarity data from different databases, including phenotypic similarity, therapeutic similarity, chemical structural similarity, and genomic similarity. Similarly, Song et al. (2019) calculated the similarity of drug pairs by 2D molecular structure similarity, 3D pharmacodynamic similarity, IPF similarity, target similarity, and ADE similarity.
Mei and Zhang (2021) proposed a simple drug target profile representation to describe drug pairs and then applied an l2-regularized logistic regression model to predict DDIs. Numerous studies have proved that data integration can introduce richer data from multiple dimensions to obtain more accurate drug characterization, thus improving the performance of drug interaction prediction. However, a large number of valuable private data from various pharmaceutical companies and academic institutions are reluctant to disclose their data due to the privacy of drug data.
To overcome the problem of private data sharing, Knott et al. (2021) proposed a privacy protection machine learning framework Crypten based on PyTorch. Crypten framework implements MPC protocol through CrypTensor object, which allows users to use automatic differentiation and neural network modules similar to PyTorch.
CONCLUSION
In this study, we propose a secure MPC-based deep learning framework for DDI predictions. This framework enhances collaboration and ensures privacy in the domain of drug discovery by employing secure MPC frameworks. By integrating diverse feature data from multiple pharmaceutical companies, deep learning operations can be conducted while maintaining the confidentiality of sensitive information. The experimental results demonstrate the outstanding performance and versatility of the proposed method across various scenarios. In conclusion, the proposed framework effectively promotes collaborative drug discovery while simultaneously preserving privacy.
Footnotes
ACKNOWLEDGMENT
The authors thank Prof. D.Q. Wei for technical discussions and for proofreading the article before submission.
AUTHORs' CONTRIBUTIONS
L.P. and X.X. conceived the original idea and developed the code for the core algorithm. S.P. designed the experiment and wrote the initial version of the article. S.L. analyzed the experimental data and edited this article. All authors reviewed and approved the final article.
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
This study was supported by NSFC Grants U19A2067; National Key R&D Program of China 2022YFC3400400; Top 10 Technical Key Project in Hunan Province 2023GK1010. The Funds of State Key Laboratory of Chemo/Biosensing and Chemometrics, the National Supercomputing Center in Changsha (![]() ), and Peng Cheng Lab.
), and Peng Cheng Lab.