
Abstract
ABSTRACT
Clinical trials indicate that the dysregulation of microRNAs (miRNAs) is closely associated with the development of diseases. Therefore, predicting miRNA-disease associations is significant for studying the pathogenesis of diseases. Since traditional wet-lab methods are resource-intensive, cost-saving computational models can be an effective complementary tool in biological experiments. In this work, a locality-constrained linear coding is proposed to predict associations (ILLCEL). Among them, ILLCEL adopts miRNA sequence similarity, miRNA functional similarity, disease semantic similarity, and interaction profile similarity obtained by locality-constrained linear coding (LLC) as the priori information. Next, features and similarities extracted from multiperspectives are input to the ensemble learning framework to improve the comprehensiveness of the prediction. Significantly, the introduction of hypergraph-regular terms improves the accuracy of prediction by describing complex associations between samples. The results under fivefold cross validation indicate that ILLCEL achieves superior prediction performance. In case studies, known associations are accurately predicted and novel associations are verified in HMDD v3.2, miRCancer, and existing literature. It is concluded that ILLCEL can be served as a powerful tool for inferring potential associations.
INTRODUCTION
Researchers have discovered that microRNAs (miRNAs) are associated with the pathogenesis of many human diseases, such as cancer, heart disease, and allergic diseases. It is estimated that each miRNA controls the expression of approximately tens of genes, and the expression of each gene is also synergistically regulated by multiple miRNAs. The development of diseases is attributed to the abnormal expression of miRNAs. In tumor cells, the translation of some mRNAs is not inhibited by the low expression of some miRNAs. Conversely, the overexpression of some miRNAs results in the strong repression of mRNAs (Deng and Huang, 2014). Thus, predicting miRNA-disease associations (MDAs) by using computational models facilitates the identification of biomarkers and the discovery of specific drugs (Cheng et al., 2005).
So far, researchers have proposed various computational methods to predict MDAs. The first type is network-based method. For example, Gu et al. (2016) presented a network consistency projection-based prediction model that predicted disease-associated miRNA without the need for negative samples (NCPMDA). Considering the complex associations between data nodes, Yu et al. (2020) constructed a three-layer heterogeneous network based on unbalanced random walk to identify MDAs. Among them, the introduction of the intermediate node lncRNA can predict the association between networks effectively (TCRWMDA). Zhong et al. (2022) proposed a prediction model named GRPAMDA, which enhanced the features of data nodes by constructing graph random propagation network and using attention network to strengthen the neighborhood features of data nodes.
The second type is matrix-based method. Matrix completion (MC) can improve prediction accuracy by making full use of known information to compensate for missing items. Therefore, Chen et al. (2021) added similarity-based neighborhood constraints to MC to recover unknown association pairs (NCMCMDA). To better preserve the experimentally validated MDAs, Zheng et al. (2022) integrated non-negative matrix factorization and MC into the prediction framework (NMFMC). Aiming to increase the sparsity, Cui et al. (2019) introduced the
Machine learning-based models that can flexibly handle large-scale data information are widely used to explore MDAs. To describe more complex relationships between samples and to fuse multiple types of information, Ding et al. (2020) combined hypergraphs and multiple kernel learning (MKL), and predicted associations using Laplace support vector machines (HGBLM). Nevertheless, the method is not sufficiently sensitive to similarity values. To preserve local information in the process of constructing similarity network, Qu et al. (2018) proposed a locally constrained linear coding (LLC) approach and used label propagation to obtain the final association scores (LLCMDA). However, the simple integration of similarities in LLCMDA and the lack of information sources affect the prediction performance.
To fuse similarity information in the miRNA (disease) space, Zhou et al. (2021) proposed a model based on MKL (DAEMKL). Among them, the features in the fused similarity networks learned by the regression model are input into the autoencoder to predict MDAs.
In this study, we design a ensemble learning model based on locality-constrained linear coding to predict MDAs (ILLCEL). The method achieves superior prediction value under fivefold cross-validation (CV) and also exhibits reliable predictive performance in case analysis. The main contributions of ILLCEL are as follows:
Compared to obtaining similarity in the single known association matrix, interaction profile (IP) similarity is calculated based on LLC to preserve local information. Considering the single priori information, the similarity features of miRNAs (diseases) are considered from different perspectives. Compared with the traditional graph structure, the introduction of hypergraph regular terms describes more complex associations between samples and improves the prediction accuracy.
MATERIALS AND METHODS
Human MDAs
The HMDD v2.0 (Li et al., 2014), which contains 5430 experimentally validated associations between 495 miRNAs and 383 diseases, is used to obtain the original association information. As shown in Equation (1), the sparse matrix
In miRNA space, IP features, sequence features, IP similarity, sequence similarity, and functional similarity are calculated to enhance priori information.
First, the associations between miRNAs and all diseases can be regarded as miRNA feature descriptors. To make full use of the known association information, miRNA feature vectors are obtained based on IP. Here, IP features extracted from the association matrix are fed into the LLC to obtain the IP similarity (Saffari and Ebrahimi-Moghadam, 2015). The specific equation is as follows:
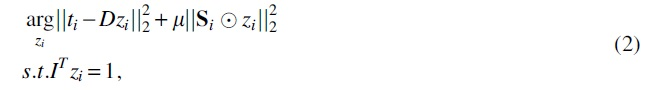
where ti is the descriptor of the i-th sample. D represents a dictionary matrix.
In the association matrix,
The Lagrangian function of Equation (2) is calculated and minimized to obtain the IP similarity
Second, since available information sources cannot directly reflect the associations between miRNAs, miRNA sequences containing attribute features are transformed into numerical vectors to characterize miRNAs. In miRBase, 495 miRNA sequence features are obtained (Kozomara and Griffiths-Jones, 2014). Next, the sequence alignment results are calculated based on the Needleman-Wunsch algorithm, and then the miRNA sequence similarity matrix
Finally, according to the similarity calculated by Wang et al. (2010), we denote the functional similarity as
We first extract the disease IP features. Then, the IP similarity based on IP features, disease semantic similarity 1, and disease semantic similarity 2 are calculated.
To begin with, IP features are obtained based on
Next, the associations between different diseases are obtained from the MeSH database (Lipscomb, 2000). Here, disease Ds is represented by Directed Acyclic Graph (DAG). In
where

Since more the parts shared between diseases, the stronger the similarity, the disease semantic similarity
where t represents the overlap part of
Finally, in the same layer of the DAG, the contribution of different disease terms to the disease semantic values may be different. Hence, the disease semantic similarity 2

In traditional graph structures, the multivariate geometric structure between data cannot be described (Zhang et al., 2018). By contrast, hypergraph learning is able to describe higher order relationships between objects. In hypergraph
The degrees of vertex and hyperedge are defined as:

Next, the diagonal matrices of dv and dh are represented by
Finally, the hypergraph Laplacian matrix
where
To improve the diversity and reliability of the model, the features and similarities considered from multiple perspectives are input into the ensemble learning. In the miRNA space,
where
Next, the
where
Considering the higher order relationships of the samples, the hypergraph-regular term is introduced. The final objective function is shown as follows:
where
The Lagrangian function of Equation (18) is expressed as follows:
After calculating the partial derivative of Equation (19),
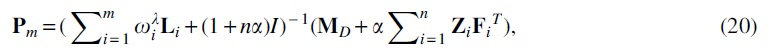
where e is a column vector and the element value of each term is 1. Taking the matrix
After obtaining the prediction matrix
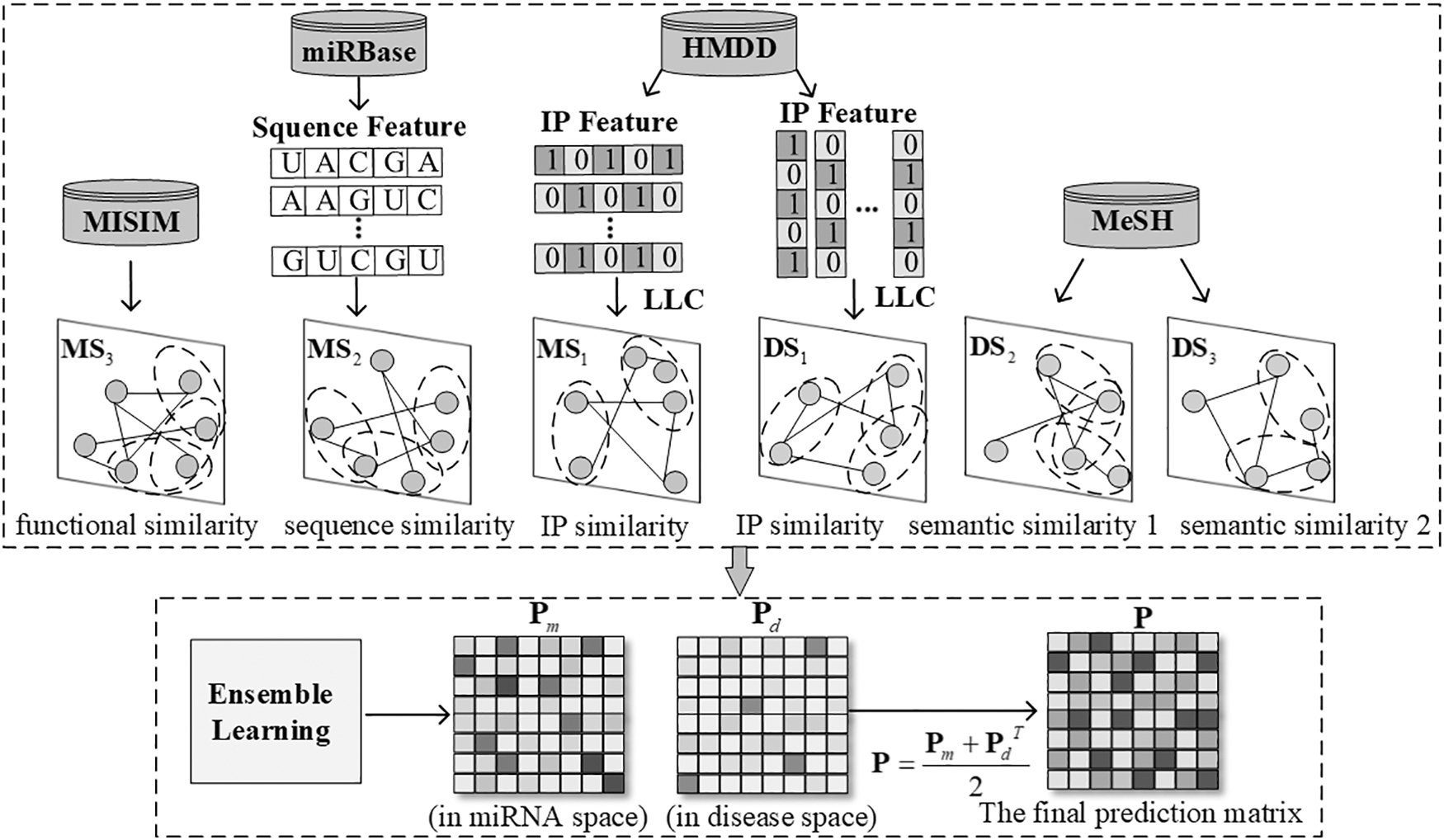
The flowchart of ILLCEL. ILLCEL, the framework of the locality-constrained linear coding-based ensemble learning model.
Performance evaluation
To test the performance of ILLCEL, we use fivefold CV. Among them, all known MDAs are randomly divided into five groups, four of which are used for training and one for testing. To avoid the bias caused by the division of samples, the average value of fivefold CV running 50 times is output. The evaluation indicator area under the curve (AUC) value, which typically ranges from 0.5 to 1, is the area under the receiver operating characteristic (ROC) curve (Fawcett, 2006). The horizontal and vertical ordinates in ROC curve denote the False Positive Rate (FPR) and True Positive Rate (TPR), respectively. The equations of FPR and TPR are shown as follows:
where TP and FP are the samples predicted to be positive. TP is actually positive and FP is negative. Besides, TN and FN are predicted to be negative, where TN actually stands for negative examples and FN actually represents positive samples.
To obtain superior prediction performance, four parameters require adjustment, that is, the regularization parameters
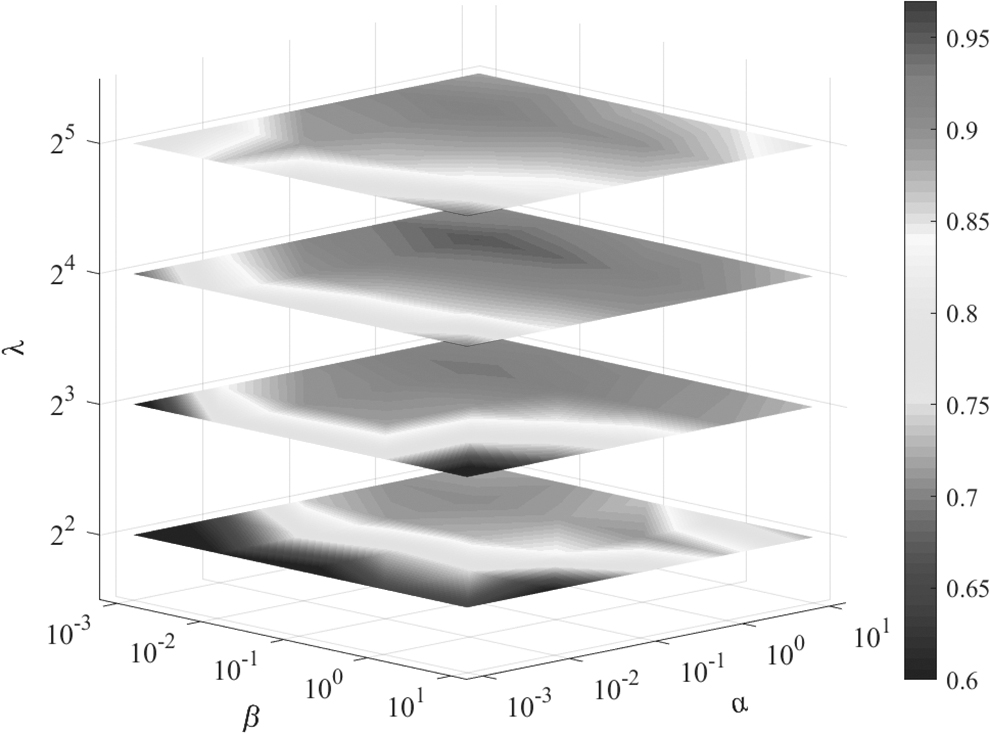
The influence of
Next, the effect of K on the prediction results in hypergraph learning is verified. Specifically, AUC values are calculated for K from 5 to 30 in steps of 5 while keeping the remaining parameters constant. As shown in Figure 3, the optimal value is obtained when
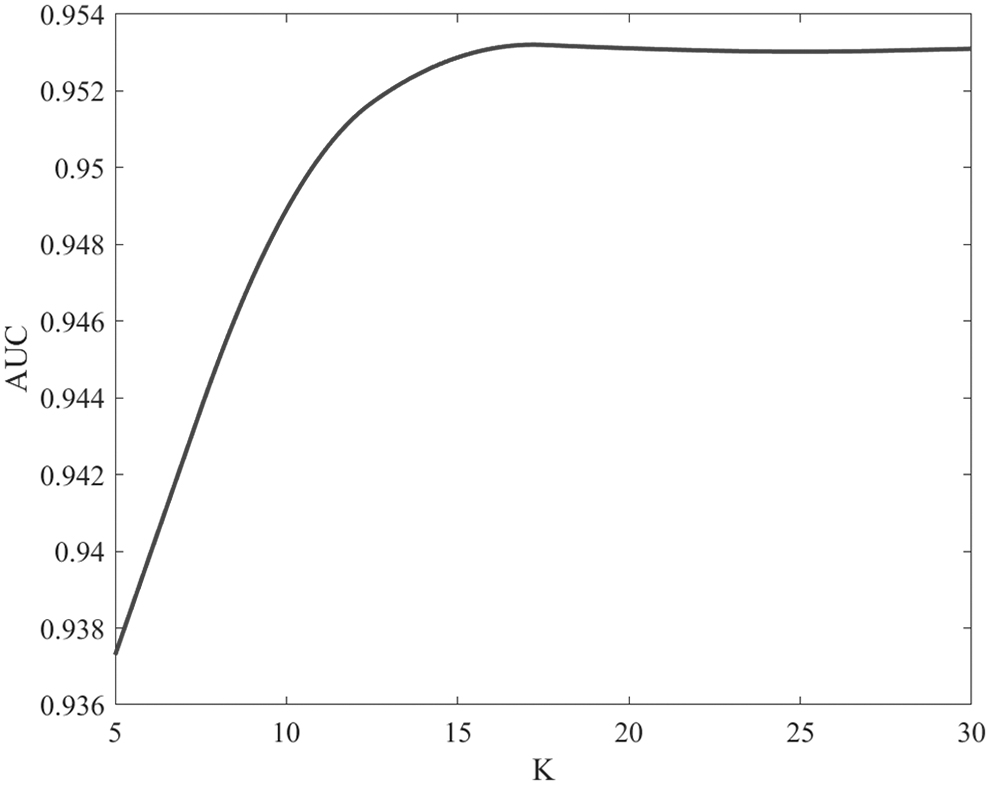
The influence of k on the prediction results.
To evaluate the prediction performance of the model, ILLCEL is utilized for comparison with NCPMDA (Gu et al., 2016), NCMCMDA (Chen et al., 2021), NMFMC (Zheng et al., 2022), TCRWMDA (Yu et al., 2020), and GRPAMDA (Zhong et al., 2022). As shown in Figure 4, the AUC values of ILLCEL, NCPMDA, NCMCMDA, NMFMC, TCRWMDA, and GRPAMDA are 0.9533, 0.8778, 0.9085, 0.9165, 0.9209, and 0.9396, respectively. It can be observed that ILLCEL has better prediction performance.
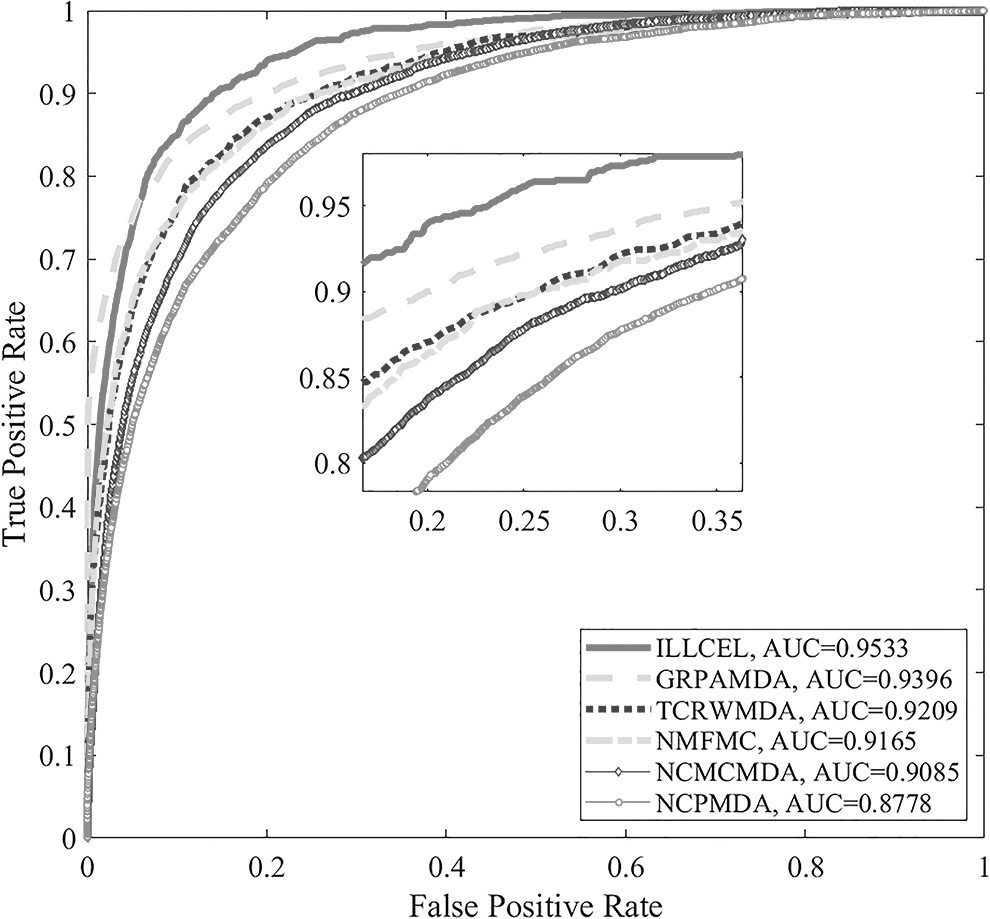
AUC values of different prediction methods. AUC, area under the curve.
Since ILLCEL performs prediction in miRNA space and disease space independently and takes the mean value as output, the predicted values on the miRNA (disease) side are compared with ILLCEL in keeping the framework consistent. As shown in Figure 5, the predicted values of our method outperform that of the single space, which indicates that more MDAs can be mined by gathering valid biological information in both spaces.
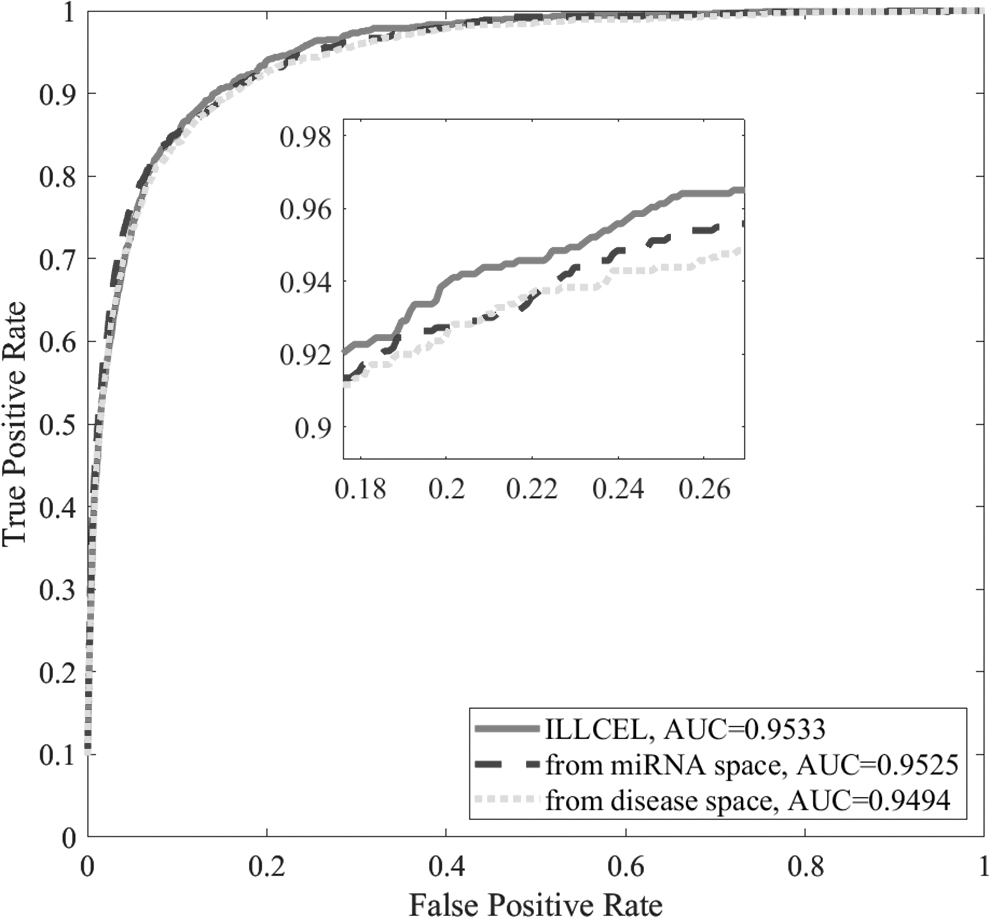
Prediction results from the miRNA (disease) side.
To further evaluate the prediction performance of ILLCEL, Kidney Neoplasms is analyzed in this section. First, the association matrix and the prediction matrix are sorted in descending order by association score to select miRNAs with higher association scores. Second, the accuracy of ILLCEL in predicting known MDAs can be demonstrated by comparing the prediction matrix with the original association matrix. Finally, HMDD v3.2 and miRCancer are used to validate the reliability of the model in predicting unknown associations. For convenience, HMDD v3.2 (Huang et al., 2019) and miRCancer (Xie et al., 2013) are denoted by H and M, respectively, in this section.
As one of the top 10 cancers in the world, renal tumor is a common urological tumor whose incidence and mortality rate are increasing year by year. Kidney neoplasms develop from various types of renal cells, such as renal cell carcinoma (RCC) and clear cell RCC (ccRCC) (Su et al., 2020). In Table 1, 9 of the top 20 miRNAs associated with kidney neoplasms have been accurately predicted. Among the remaining 11 predicted miRNAs, miR-200a, miR-155, miR-375, and miR-203 can be confirmed in H or M. It has been indicated that miR-429 can inhibit cancer cell production by targeting AKT1 in RCC.
Predicted MicroRNAs for Kidney Neoplasms
Predicted MicroRNAs for Kidney Neoplasms
In addition, the growth and migration of RCC are inhibited by the dysregulation of miR-99a (Cui et al., 2012) and miR-200b (Li et al., 2019). Significantly, miR-20a can be used as a biomarker for identifying RCC (Oto et al., 2021). The expression of miR-9 (Xie et al., 2018) tends to be downregulated in ccRCC compared to normal renal tissue, while the expression levels of miR-125b (Fu et al., 2014) and miR-146a (Ichii et al., 2012) are enhanced.
As a type of intracranial tumor, brain neoplasms arise from the uncontrolled growth of abnormal cells. Among them, medulloblastoma (MB) is the most common malignant brain tumor in children. It has been shown that miR-9 can exert a great effect on MB by regulating HES1. Furthermore, the expression of miRNA target genes is positively associated with the accumulation of miRNAs in the cerebellum (Dubuc et al., 2012). In Table 2, the top 20 miRNAs associated with brain neoplasms are selected. Among them, 11 known associations have been successfully confirmed. In addition, 4 novel predicted associations can be found in H or M. It has been documented that the development of gliomas is closely associated with the dysregulation of miR-125b, miR-92a, and miR-145. In glioblastoma, miR-29a can exert a significant inhibitory effect on cells by regulating the platelet-derived growth factor (PDGF) pathway (Yang et al., 2019).
Predicted MicroRNAs for Brain Neoplasms
Currently, identifying that miRNAs associated with complex diseases is a critical research topic in the biomedical field. In this article, a ensemble learning model based on locality-constrained linear coding is proposed to predict associations (ILLCEL). Specifically, features and similarities from different bioinformatics sources are fed into the ensemble learning. To preserve the local information, LLC converts the IP feature descriptors extracted from the known association matrix into an encoding matrix. Considering the high-order relationships between samples, the hypergraph-regular term is introduced into the model. After integrating the prediction matrices in miRNA space and disease space, unknown associations are identified. The AUC value of ILLCEL under fivefold CV is 0.9533, which is superior to the comparison methods. In the case studies, known associations have been accurately predicted, and most of the novel associations are confirmed.
However, ILLCEL still has some shortcomings. First, the multisource bioinformation is not reasonably integrated when inputted into the ensemble learning, which may affect the prediction accuracy. Second, a more reasonable strategy needs to be designed to select the optimal combination of parameters.
Footnotes
AUTHORs' CONTRIBUTIONS
Y.S.: writing-review and editing. J.-X.L.: review and editing. J.W.: data collection. Y.-L.G.: visualization. B.-X.G.: formal analysis.
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
This work was supported by the National Natural Science Foundation of China (Grant No. 62172254).