
Abstract
Molecular prediction tasks normally demand a series of professional experiments to label the target molecule, which suffers from the limited labeled data problem. One of the semisupervised learning paradigms, known as self-training, utilizes both labeled and unlabeled data. Specifically, a teacher model is trained using labeled data and produces pseudo labels for unlabeled data. These labeled and pseudo-labeled data are then jointly used to train a student model. However, the pseudo labels generated from the teacher model are generally not sufficiently accurate. Thus, we propose a robust self-training strategy by exploring robust loss function to handle such noisy labels in two paradigms, that is, generic and adaptive. We have conducted experiments on three molecular biology prediction tasks with four backbone models to gradually evaluate the performance of the proposed robust self-training strategy. The results demonstrate that the proposed method enhances prediction performance across all tasks, notably within molecular regression tasks, where there has been an average enhancement of 41.5%. Furthermore, the visualization analysis confirms the superiority of our method. Our proposed robust self-training is a simple yet effective strategy that efficiently improves molecular biology prediction performance. It tackles the labeled data insufficient issue in molecular biology by taking advantage of both labeled and unlabeled data. Moreover, it can be easily embedded with any prediction task, which serves as a universal approach for the bioinformatics community.
INTRODUCTION
Molecular prediction tasks are essential and fundamental within bioinformatics fields such as drug discovery (Ching et al., 2018; Guo et al., 2020a; Paul et al., 2010), which contain various molecule-relevant tasks, including molecular property prediction and protein secondary or tertiary structure prediction. With the advance of deep learning techniques, more and more studies address these tasks with various deep learning models (Guo et al., 2022; Heffernan et al., 2015; Ma et al., 2022a; Wang et al., 2016; Wu et al., 2018; Xu et al., 2018; Yang et al., 2019). These prediction tasks are well known as supervised problems, where labeled data serve as input and computational models are employed to predict the corresponding labels.
Many existing methods target such problems in this manner (Duvenaud et al., 2015; Gilmer et al., 2017; Guo et al., 2021; Ma et al., 2021; Ma et al., 2020a; Sønderby and Winther, 2014; Wu et al., 2018). However, one major challenge in molecular biology is that labeled data are limited and also difficult to obtain. The process usually involves a series of professional experiments, which can be both time-consuming and costly. Therefore, more paradigms have been developed to take advantage of unlabeled data to assist supervised learning, such as semisupervised learning (Hu et al., 2019; Mann and McCallum, 2007; Rong et al., 2020; Wang et al., 2019a; Zhang et al., 2018; Zhao et al., 2014). Within this field, a simple yet effective paradigm that leverages both unlabeled and labeled data, known as self-training, is rarely explored for molecular biology prediction tasks.
Self-training is generally established in four steps: (1) A teacher model is trained using labeled data; (2) the trained teacher model is subsequently utilized to generate pseudo labels for unlabeled data; (3) the labeled data and the pseudo-labeled data are combined to train a student model; and (4) the student model then becomes the teacher model to repeat steps 2–3 until the training is converged. This approach allows more data to be included in the training process and enables the student model to inherit from the teacher. This paradigm is easy to implement and effective to boost the training process. Self-training has been widely used in other areas and obtained promising performance, for example, Computer Vision (CV) (Babakhin et al., 2019; Xie et al., 2020; Zoph et al., 2020) and Nature Language Processing (NLP) (Cheng, 2019; He et al., 2019; Li et al., 2019).
Notably, it has achieved significant success in image classification area. Xie et al. (2020) propose a Noisy Student model, which constructs a larger student model by adding noise to force the student to learn more complex information. Touvron et al. (2021) explore the attention mechanism and develop a transformer-based model that follows the teacher-student flow. Meta-Pseudo Labels (Pham et al., 2021) proposes to dynamically update the teacher model based on the student performance for more effective learning from limited labeled data.
A key reason for the success is that not only are the unlabeled data enormous but also the size of labeled data is quite large, and so are the training models. Thus, the teacher model can sufficiently learn from the labeled data and achieve favorable performance, from which the student can then learn further. Take image classification as an example, the teacher model is trained on the ImageNet dataset, which contains over 14 million images with hundreds of millions of parameters. The prediction performance can achieve over 85% for top-1 accuracy and 95% for top-5 accuracy in a 1000-class prediction problem (Xie et al., 2020).
However, for most molecular biology prediction tasks, the size of the labeled dataset typically amounts to only a few thousand, with the corresponding prediction performance not as high as in image classification. Such scenarios lead to a problem: The generated pseudo labels may not be accurate. Such noisy labels may further bias student learning. Therefore, addressing label noise is the major concern when establishing self-training in the field of molecular biology.
One straightforward way to encourage the model to learn from the noisy labels is to design a loss with regularization to leverage the neural network learning. Mean absolute error (MAE) and cross-entropy (CE) loss are two commonly used loss functions in prediction tasks. MAE is typically employed in regression tasks and CE for classification tasks. While MAE has been theoretically proven to be robust to label noise during the training, the CE is not (Ghosh et al., 2017). Recently, robust loss functions have been studied to tackle the noisy label problem in classification tasks by generalizing MAE and CE, and have achieved impressive performance in image classification tasks (Englesson and Azizpour, 2021; Ghosh et al., 2017; Ma et al., 2020b; Wang et al., 2019b; Zhang and Sabuncu, 2018).
In this article, we leverage robust loss function and self-training to form a robust self-training framework for molecular biology prediction tasks in two paradigms, that is, generic and adaptive. Extensive experiments have been conducted over molecular regression and classification tasks, as well as protein secondary structure prediction (PSSP) tasks, to gradually evaluate the effectiveness of the proposed method.
Our contributions can be summarized as follows: (1) We propose a robust self-training paradigm that utilizes robust loss to constrain the student training; (2) the proposed framework is straightforward and easy to fit into any prediction task, which is a simple yet practical strategy to promote molecular prediction tasks; (3) the adaptive paradigm further specifies and enhances the training on unlabeled data; and (4) extensive experiments on various molecular prediction tasks demonstrate that self-training can improve the prediction performance by involving more unlabeled data, and the robust loss can further boost the performance by leveraging the label noise, as well as alleviating the overfitting during the training.
MATERIALS AND METHODS
Problem definition
Molecular biology prediction problems can be further referred to as regression problems or classification problems. Given a molecule
Graph-based models generally take the molecular graph structure as the input, and generate a continuous vector
Robust self-training overview
Our proposed robust self-training strategy is implemented on top of the self-training framework. Figure 1 illustrates the overall architecture, which can be viewed as two parts, train teacher and train student. First of all, a teacher model is trained on the labeled dataset
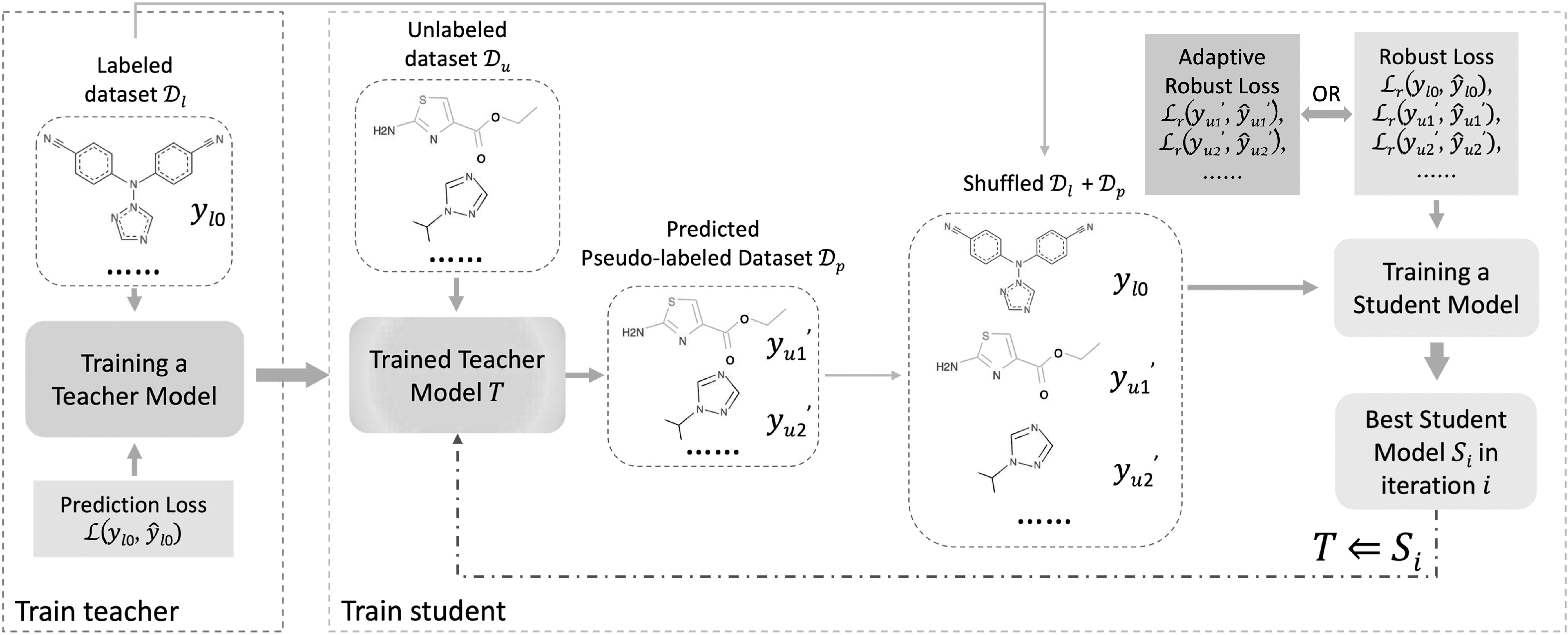
An overview illustration of the robust self-training architecture. More details are described in Section 2.2.
After that, the student training process begins. It starts with generating the pseudo labels for the unlabeled dataset
Moreover, we design an adaptive robust self-training paradigm to further enhance the student training, which is illustrated as the adaptive option in Figure 1 and Algorithm 1. Specifically, robust loss
Molecular property prediction task can be considered as two parts in view of deep learning, which are molecular encoder model and prediction model. Molecular encoder model generates a vector that represents the input molecule, and the prediction model takes the vector to make a prediction. In our experiments, the backbone models of the regression and classification task are graph based. We give a universal definition here for the graph-based encoder and the prediction model.
Molecule
GNN-based models generally perform a message passing and state update protocol. In specific, the state of the target atom v at step d (

where
After going through the graph encoder model, the graph representation
where
MAE loss is theoretically proved to be robust (Ghosh et al., 2017). Therefore, we first conduct experiments on molecular regression tasks with MAE as the loss function. EGNN (Satorras et al., 2021) is one of the most recent works to address such problems. Other than the commonly used message-passing process based on the graph structure and features, EGNN further explores the geometric information by considering the atom coordinates
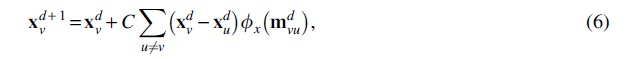
where
MAE is used as the robust loss function to constrain the network training with regard to noisy labels, which is defined as follows:

where M is the size of the dataset.
Classification tasks are dominating for molecular property prediction problems as well. We then conduct experiments over classification tasks to evaluate the effectiveness of the self-training paradigm. However, the commonly used CE loss is not robust, so we employ the generalized cross-entropy (GCE) loss (Zhang and Sabuncu, 2018) to boost the self-training. In particular, GCE loss is employed to replace the CE loss during the student training phase, where the unlabeled data are predicted with pseudo labels.
Furthermore, we extend this strategy to an adaptive version, where the GCE loss is calculated and back-propagated solely for the unlabeled data that have the generated pseudo labels from the teacher model. The backbone model utilized for this task is GIN (Xu et al., 2018). GIN is theoretically proved as one of the most powerful GNN models. It utilizes MLP for state update, and employs a concatenate operation over all passing steps during the readout phase. The updated rule in Equations (2) and (3) can be summarized as follows:

where
GCE loss is a generalized version of CE and MAE. The CE loss is defined by

for a K-class classification problem (K = 2 for binary classification), where yk is the one-hot encoding label, and
GCE loss is reduced to CE loss and MAE loss when
To further prove the universality and effectiveness of our method, we conduct experiments with two state-of-the-art PSSP methods as the backbone models to evaluate proposed robust self-training strategy.
AWD-GRU (Moffat and Jones, 2021) is one recent work that utilizes Gated Recurrent Unit (GRU) for PSSP. It exploits three layers of GRUs, where each layer GRU with DropConnect (Wan et al., 2013) is applied to the hidden-to-hidden weight matrices.
EnsembleASP (Guo et al., 2020b) adopts CNN-based networks to capture local context features and bidirectional-LSTM (bidirectional-Long Short-Term Memory) for long-term dependencies. Moreover, the ASP network (a modified version of Atrous Spatial Pyramid Pooling network) (Chen et al., 2018) further improves the prediction performance by identifying the boundary of successive amino acids that have the same secondary structure. In this work, we use single protein sequence without profile information as the input.
Both the backbone models address the three-state secondary structure prediction problem, which forms a three-class classification problem. The robust loss exploited here follows the same implementation as in Equations (11) and (12) to make our experiments consistent:
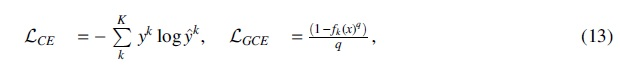
where K equals to 3, and
RESULTS
Extensive experiments are conducted gradually to evaluate the effectiveness of the proposed robust self-training strategy. We first implement self-training on molecular regression tasks with MAE loss as the robust loss function. Then we explore GCE loss on the molecular classification tasks. Finally, we establish self-training with GCE loss on a large-scale protein secondary structure dataset to further demonstrate the superiority of the proposed method. Source code will be released soon after cleaning up.
Dataset description and setup
Molecular property datasets
QM9 (Ramakrishnan et al., 2014) is a standard benchmark for molecular property regression problems. It is a subset of GDB-17 database (Ruddigkeit et al., 2012), which contains 134k molecules. It comprehensively provides 12 quantum chemical properties for each molecule, including geometric, energetic, electronic, and thermodynamic. HIV is introduced by the Drug Therapeutics Program (DTP) AIDS Antiviral Screen. It contains the test result of 41,127 molecule compounds with the ability for inhibiting HIV replication. The widely used version provided by MoleculeNet (Wu et al., 2018) contains inactive labels and active labels, which makes it a binary classification task.
For all molecular property prediction tasks, we randomly select 50% of the data as the unlabeled dataset, and the rest is used as the labeled dataset with a 3:1:1 training/validation/test ratio. We do not use an external unlabeled dataset here since most molecules may not express target property at all, which may lead to a biased comparison. Therefore, we further conduct experiments on PSSP since every protein contains the secondary structure labels.
Protein secondary structure datasets
For PSSP, three datasets are used in total: labeled dataset, testing dataset, and unlabeled dataset.
Labeled dataset construction contains two steps. First, the protein sequence is generated by Pisces server (Wang and Dunbrack, 2003) with a maximum consistency of 70% between structures, a maximum resolution of 2.6A (Moffat and Jones, 2021), and a length not exceeding 700. The resulting labeled dataset contains 25,503 protein sequences, and is split into training and validation datasets with a 95:5 ratio. Next, the corresponding 3-state secondary structure labels are generated by DSSP (Kabsch and Sander, 1983).
Testing dataset utilized in the experiments is not selected from the labeled dataset, since the widely used dataset to test PSSP is CB513 (Cuff and Barton, 1999). Thus, we follow the standard protocol to evaluate the prediction performance on CB513, which contains 513 proteins generated from Zhou and Troyanskaya (2014).
Unlabeled dataset is generated using Uniclust30 (Mirdita et al., 2017), which consists of UniProtKB (Uniprot Consortium, 2019) sequences clustered to 30% identity, and the length is less or equal to 700. We remove those protein sequences that share homology information with the labeled dataset, and then use the remaining 201,408 protein sequences as our unlabeled dataset.
To prevent data leakage, we perform strict screening of the super homologous family information. CATH (Sillitoe et al., 2021) is used for homology assessment, where any sequence overlapped with the testing dataset (CB513) at the superfamily level is removed from both unlabeled dataset and labeled dataset by cross-referencing.
It is noteworthy that for each task, the validation and test datasets are fixed after construction, and remain the same for all the comparison experiments. The unlabeled dataset is only involved in the training phase for the self-training procedure.
Experimental details
Baselines
For all the experiments, we consider training solely on the labeled datasets as the fundamental baselines, which is denoted as “-labeled.” Then, we establish our vanilla implementation by running experiments with self-training paradigm on both labeled dataset and unlabeled dataset, denoted as “-self-training.” Finally, we integrate robust loss with our vanilla self-training benchmark to demonstrate the superiority of our robust self-training, denoted as “-robust.” For the further enhanced adaptive paradigm with both original loss and robust loss, the experiments are denoted as “-adaptive-robust.” Note that the adaptive option is only established on the classification tasks since MAE loss is sorely used in the regression tasks. For molecular property prediction, since the unlabeled dataset is formed by randomly selecting 50% from the original labeled dataset, we also compare the performance when using the original backbone model without self-training on all the data with labels, denoted as”-all.”
Overall, extensive experiments are conducted over three tasks with four backbone models: EGNN for molecular regression task, GIN for molecular classification task, and AWD-GRU and EnsembleASP for PSSP task.
Evaluation metric
We follow the commonly used evaluation metric for each task. In specific, MAE is used as the evaluation criteria for molecular regression tasks on QM9; area under the receiver operating characteristic curve (ROC-AUC) and Precision-Recall Area Under Curve (PRC-AUC) are used for molecular classification tasks; Q3 accuracy is used for protein three-state secondary structure prediction (Moffat and Jones, 2021).
Configurations
We follow the original implementation and settings of the backbone models, and implement robust self-training on top of them. All the hyperparameters of the backbone models remain the same to ensure a fair comparison. For the settings of robust self-training, we perform three iterations for the student training, and tune the hyperparameter q when employing GCE loss. For molecular classification task, we run the experiments three times to alleviate the randomness since HIV dataset is much smaller than other datasets, leading to relatively unstable performance. We take the average and standard deviation of the evaluation scores as the final results.
For molecular regression and PSSP tasks, we follow the original configurations and evaluations to run the experiments one time. The results do not vary much since the training data are sufficiently large and the converged stage is stable.
Training strategy
We follow the same procedure for all three tasks. First, we train a teacher model on the labeled data, and use it to generate pseudo labels for the unlabeled dataset. Next, for the vanilla self-training, we train the student model, which takes the teacher model as the initialization on the combined labeled and pseudo-labeled dataset. Note that the pseudo-labeled dataset is only merged into the training dataset along with the labeled training dataset.
The validation and test datasets remain the same from the teacher model training. Furthermore, we choose the best student model in the current iteration as the new teacher model to generate a new pseudo-labeled dataset and initialize the student model for the next iteration. We run the student training for three iterations, and take the best validation model to evaluate the test dataset performance. For robust self-training, the procedure is the same as vanilla self-training, except robust loss function is employed. Similar for adaptive robust self-training, where the robust loss is used for unlabeled data and the original loss is used for labeled data.
Experimental results
Molecular property prediction
Our first experiment is to employ the self-training paradigm directly on molecular regression tasks, since MAE is theoretically robust to noisy labels. The comparison results for each property are shown in Table 1. “EGNN-labeled” represents the baseline model, which is trained on the labeled datasets. “EGNN-self-training” denotes the experiments that perform self-training by including the unlabeled datasets.
Mean Absolute Error for Each Molecular Property Regression Benchmark on QM9 Dataset
Mean Absolute Error for Each Molecular Property Regression Benchmark on QM9 Dataset

Since the inherently used MAE is theoretically proven to be a robust loss, “EGNN-self-training” is equivalent to our proposed paradigm. “EGNN-all” represents the scenario of using the original backbone model, without self-training, on all the data with labels. As we can observe, the performance of the self-training strategy outperforms EGNN-label consistently by a 41.5% average improvement. Moreover, the performance is competitive against the supervised training on the all-labeled dataset. Our implementation achieves the best performance on 9/12 tasks compared with the original EGNN-all on all 134k labeled data, which gains the average MAE boost by 7.2%. The experiments on regression tasks with MAE sufficiently demonstrate that robust loss function is a perfect fit for self-training strategy by dealing with the generated pseudo labels.
We then conduct experiments on the HIV dataset to evaluate how self-training performs on classification tasks. As shown in Figure 2, the improvement of directly implementing self-training is limited, which is reasonable since CE loss is not theoretically robust (Ghosh et al., 2017). Therefore, we explore robust loss function GCE and integrate it with self-training to form the robust self-training paradigm, which further boosts the ROC-AUC to 0.822. The adaptive paradigm, which employs robust loss on unlabeled dataset, further improves the performance to 0.826. Moreover, our methods are competitive with the original GIN implementation on the all-labeled dataset with a 0.002–0.006 improvement. Note that in our self-training experiments, 50% of the dataset is treated as unlabeled, while GIN-all is trained on 100% labeled dataset.
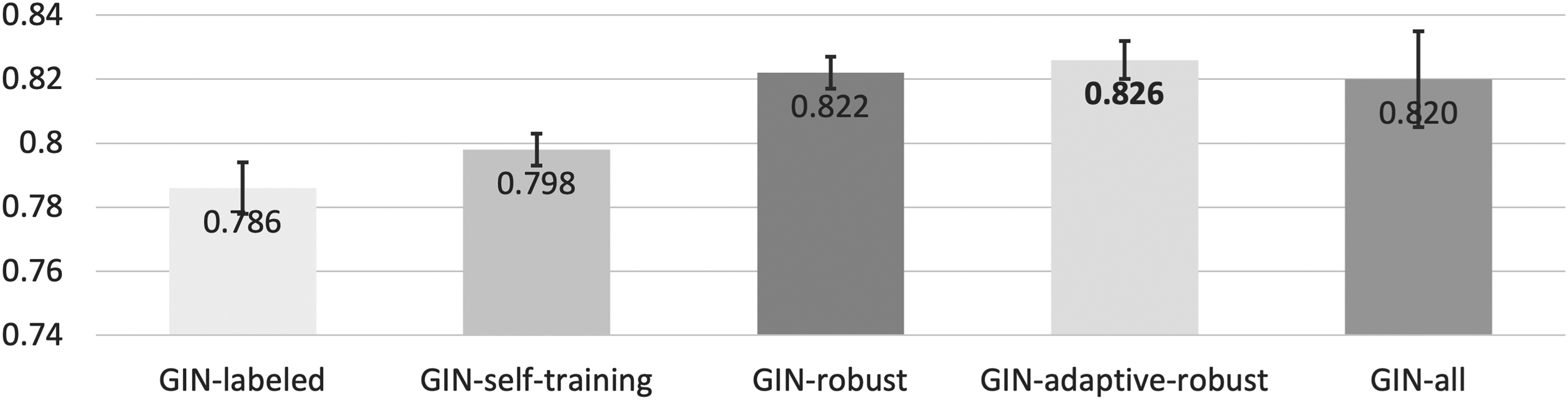
ROC-AUC score for molecular property classification benchmark on HIV dataset. Higher score is better. ROC-AUC, area under the receiver operating characteristic curve.
To further evaluate the performance of our proposed robust self-training paradigm, we conduct experiments on more diverse datasets and also include the PRC-AUC score as an evaluation metric. The results are shown in Table 2. “GIN-labeled” represents training on the labeled dataset, which also serves as the teacher model. “Ours” represents the performance of our proposed adaptive robust self-training framework. “GIN-labeled-RL” denotes the teacher model trained with robust loss instead of the original CE loss.
Dataset Information and Comparison Experiments on HIV, Tox21, Toxcast, and MUV Datasets
ROC denotes the ROC-AUC score, and PRC is the PRC-AUC score. Higher value indicates better performance. Best scores are marked in bold.
GIN, Graph Isomorphism Network; PRC-AUC, precision-recall area under curve; RL, robust loss; ROC-AUC, area under the receiver operating characteristic curve.
As observed, our models consistently improve upon the teacher model in both ROC-AUC and PRC-AUC scores. The performance of “GIN-labeled-RL” is not as good, which is reasonable since the original dataset is labeled and does not include much noise. Therefore, the benefits of robust loss are not fully realized. In addition, Tox21, Toxcast, and MUV are multitask datasets from MoleculeNet (Wu et al., 2018), containing more than one property label with missing labels, which could introduce additional challenges during training.
We also compare and visualize the ROC-AUC scores from training, validation, and testing between GIN-self-training, GIN-robust, and GIN-adaptive-robust on HIV dataset, as shown in Figure 3. GIN-self-training represents the vanilla self-training, GIN-robust illustrates self-training with robust loss, and GIN-adaptive-robust denotes the adaptive paradigm. The teacher model is trained for 150 epochs, so is for the student training during each iteration.
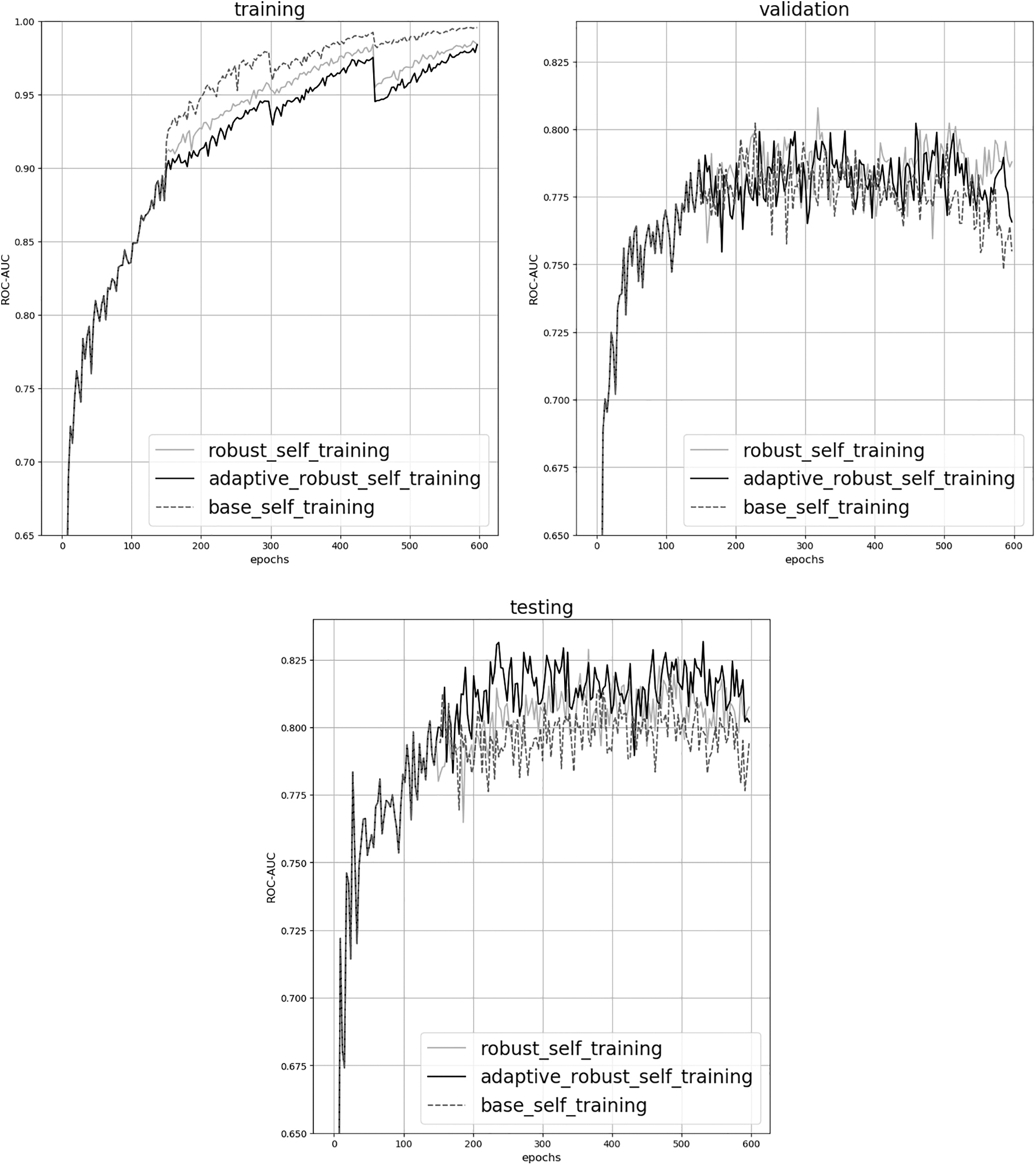
ROC-AUC visualization of training, validation, and testing on HIV dataset. X-axis is the epoch number, and Y-axis is the ROC-AUC value. Dashed lines represent GIN-self-training, darker solid lines indicate GIN-robust, and lighter solid lines denote GIN-adaptive-robust. GIN, Graph Isomorphism Network.
The curves are overlapped for the first 150 epochs since we load the same teacher model to make a fair and explicit comparison of how the robust loss function promotes self-training. From the training curves, we can clearly observe re-training progress between every 150 epochs, and the ROC-AUC values of GIN-robust (blue lines) and GIN-adaptive-robust (orange lines) are much more stable than GIN-self-training (green dashed lines). Meanwhile, as shown in both training and validation curves, GIN-self-training gets easily overfitted compared with our robust self-training methods. Furthermore, the test curves demonstrate that with the help of robust loss, the student model can keep learning better during iteration training.
To mimic the real-world scenario, which means utilizing data that actually does not have a label, we conduct experiments over PSSP. Unlike molecular property prediction, where the external unlabeled dataset may not contain any molecules that express the target property, every protein molecule certainly contains secondary structure labels. Therefore, we use a large-scale external unlabeled dataset to run our robust self-training experiments. As shown in Figure 4, the prediction performance is as expected. PSSP-self-training slightly improves the performance by using self-training, and then along with robust loss, PSSP-robust further boosts the Q3 accuracy to 0.734 and 0.735 for the two backbone models, respectively.

Q3 accuracy for the PSSP task. Higher score is better. PSSP, protein secondary structure prediction.
It is noteworthy that the performance of PSSP-adaptive-robust is not as good as PSSP-robust. The reason is that for PSSP, the theoretical limit of accuracy has been estimated to be 92% (Dill and MacCallum, 2012; Ho et al., 2021) due to the potential existence of noise in the observation of secondary structures. Consequently, the labeled data used in our self-training framework may already contain noise.
We have conducted experiments that substitute the original CE loss on the PSSP-labeled dataset with robust loss, denoted as PSSP-labeled-robust. We can observe that the performance is competitive with PSSP-labeled, which indicates that the labeled data might include noisy labels. Therefore, directly utilizing robust loss for the entire dataset is the optimal strategy for such a scenario, as revealed by the comparison results. It is empirically demonstrated that the proposed robust self-training is a simple yet effective strategy for molecular biology prediction tasks by sufficiently utilizing both unlabeled and labeled data.
Extensive experiments gradually demonstrate the superiority of proposed robust self-training. First, the regression tasks associated with MAE loss have achieved remarkable improvement. Since MAE is theoretically proven to be robust, the proposed method that integrates robust loss with self-training has been proven effective. Next, we conduct more experiments using self-training with robust GCE loss to evaluate the performance on classification tasks.
The results confirm that self-training can slightly improve the prediction performance, and with the help of robust loss, the training becomes more stable and the performance is further boosted by a large margin. Moreover, considering that noisy labels in the real world are difficult to recognize, while real label data (labeled data) and data containing noisy labels (pseudo-labeled data) can be distinguished in the self-training process. We design the adaptive paradigm that conditionally employs robust loss, that is, keep the original classification loss for the labeled data, while utilizing robust loss for the pseudo-labeled data. The results further confirm the effectiveness of the robust loss in handling noisy labels.
Our study represents a preliminary step toward this robust self-training paradigm, within which several unexplored directions remain to be further studied. For example, self-training with MAE works great since the default MAE loss for regression tasks is robust. Nevertheless, for classification tasks, robust loss like GCE is a generalized version between CE and MAE, which can be considered a tradeoff between robust loss and traditional classification loss. Other forms of robust loss may be more suitable for different prediction tasks.
Moreover, the Alternative Convex Search (ACS) algorithm is introduced to optimize a truncated version of the GCE loss by balancing the weights and model parameters. Given that molecular prediction tasks often involve complex, multidimensional data with unique challenges such as label imbalance, multilabel outputs, and pseudo-labeling, such optimization may not yield optimal results in naive settings. Further and deeper studies can be explored by deploying more rigorous algorithms to adapt them to the specific demands of molecular prediction tasks.
CONCLUSIONS
In this study, we propose a robust self-training paradigm for molecular prediction tasks by exploring robust loss functions to constrain the self-training process. Extensive experiments over molecular regression, molecular classification, and PSSP tasks have demonstrated that self-training accompanied by robust loss can boost prediction performance by taking advantage of both labeled and unlabeled data. Moreover, our proposed robust self-training is model and task agnostic, which can be easily inserted into any molecular biology prediction task, and benefits the general computational molecular biology society.
Footnotes
ACKNOWLEDGMENT
AUTHORs' CONTRIBUTIONS
H.M.: Conceptualization and methodology; F.J.: Formal analysis; Y.R.: Resources; Y.G.: Writing—review and editing; and J.H.: Supervision.
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no conflicting financial interests.
FUNDING INFORMATION
This work was partially supported by the Cancer Prevention and Research Institute of Texas (CPRIT) award (RP230363).