
Abstract
The single-matrix amino acid (AA) substitution models are widely used in phylogenetic analyses; however, they are unable to properly model the heterogeneity of AA substitution rates among sites. The multi-matrix mixture models can handle the site rate heterogeneity and outperform the single-matrix models. Estimating multi-matrix mixture models is a complex process and no computer program is available for this task. In this study, we implemented a computer program of the so-called QMix based on the algorithm of LG4X and LG4M with several enhancements to automatically estimate multi-matrix mixture models from large datasets. QMix employs QMaker algorithm instead of XRATE algorithm to accurately and rapidly estimate the parameters of models. It is able to estimate mixture models with different number of matrices and supports multi-threading computing to efficiently estimate models from thousands of genes. We re-estimate mixture models LG4X and LG4M from 1471 HSSP alignments. The re-estimated models (HP4X and HP4M) are slightly better than LG4X and LG4M in building maximum likelihood trees from HSSP and TreeBASE datasets. QMix program required about 10 hours on a computer with 18 cores to estimate a mixture model with four matrices from 200 HSSP alignments. It is easy to use and freely available for researchers.
INTRODUCTION
The amino acid (AA) substitution models are used to analyze protein sequences. The AA substitution process is normally described by a Markov process with properties of time-reversible, time-continuous, time-homologous, and stationary (Durbin et al., 2006). Popular AA substitution models consist of one replacement matrix representing the substitution rates among amino acids for all sites (Minh et al., 2021). The evolution of amino acids varies among sites and depends on many factors such as genetic codes, solvent accessibility, and protein functions (Le et al., 2012). The single-matrix model simply uses a discrete gamma distribution to model the heterogeneity of evolutionary rates (Yang, 1993); however, it is incapable of modeling the variability of substitution patterns among sites. For example, buried sites and exposed sites correspond to slow and fast substitution patterns and require very different AA replacement matrices. Using several replacement matrices, each for one rate category is a better approach (Le et al., 2012).
Two 4-matrix mixture models LG4X and LG4M have been estimated from 1471 HSSP alignments (Le et al., 2012). The LG4M model has four replacement matrices each corresponding to one rate category of the discrete gamma distribution (Yang, 1993). The LG4X model follows a distribution-free scheme for the site rates (Yang, 1995). The multi-matrix models outperform the single-matrix models in inferring maximum likelihood trees while requiring the same memory space and similar running times (Le et al., 2012). The multi-matrix model consists of more parameters than the single-matrix model, therefore, estimating a multi-matrix model is more complicated, more time-consuming, and requires larger datasets.
The workflow to estimate LG4X and LG4M is very complicated for biologists (even for bioinformaticians); and not yet implemented as a computer program. To overcome the burden for researchers, we implemented the workflow in QMix program with several improvements to automatically estimate multi-matrix models from large datasets.
METHODS
The training dataset used to estimate an amino acid substitution model include N alignments denoted by
Optimizing
The QMix program consists of four main steps to iteratively optimize Initial step: Initialize Categorizing step: Estimate trees Estimating parameters: Estimate Stopping step: Compare new matrices of
We evaluated the performance of both single-matrix and multi-matrix models in inferring maximum likelihood trees. As models might have different number of free parameters (e.g., multi-matrix models have more parameters than single-matrix models), we used AIC criteria (Akaike, 1974) to assess the performance of the models, i.e.,
RESULTS
QMix validation
We employed the HSSP dataset that was used to estimate four-matrix mixture models LG4M and LG4X (Le et al., 2012) to validate the QMix program. The dataset comprises 1771 alignments containing about 27 million amino acids (on average, each alignment consists of about 56 sequences and 254 sites). The dataset was divided into two parts: the training part of 1471 alignments and the testing part of 300 remaining alignments. The training and testing parts in this study were the same as those used in the article of LG4M and LG4X models (Le et al., 2012). We employed QMix to estimate four-matrix mixture models HP4M (rate categories follow a discrete gamma distribution) and HP4X (rate categories follow a rate distribution-free scheme) from the 1471 training. To evaluate the stability of QMix, we also estimated two 4-matrix models HP4X.200 and HP4M.200 from 200 HSSP alignments.
Table 1 shows that matrices for the “medium” and “fast” rates of the models have high correlations. The matrices for “very slow” and “slow” rates have lower correlations. The “very slow” and “slow” matrices of LG4X have considerably low correlations with those of other matrices. The deviation of “very slow” and “slow” matrices of LG4X is perhaps due to the sensitivity of XRATE algorithm with starting parameter values as noted by the authors (Le et al., 2012). We observe high correlations between matrices of HP4X and HP4X.200; and reasonable correlations between matrices of HP4M and HP4M.200. The results indicate the stability of the QMix program.
The Pearson Correlations Between Matrices of LG4X, LG4M, HP4X, HP4M, HP4X.200, HP4M.200 Mixture Models
The Pearson Correlations Between Matrices of LG4X, LG4M, HP4X, HP4M, HP4X.200, HP4M.200 Mixture Models
Figure 1 shows the relative difference between exchangeability coefficients in matrices of HP4X and LG4X. The “fast” matrices of HP4X and LG4X are highly similar with only two rates that show five times difference, while the “very slow” matrices are more different (36 rates show at least five times difference). We observed similar results when comparing matrices of HP4M and LG4M.
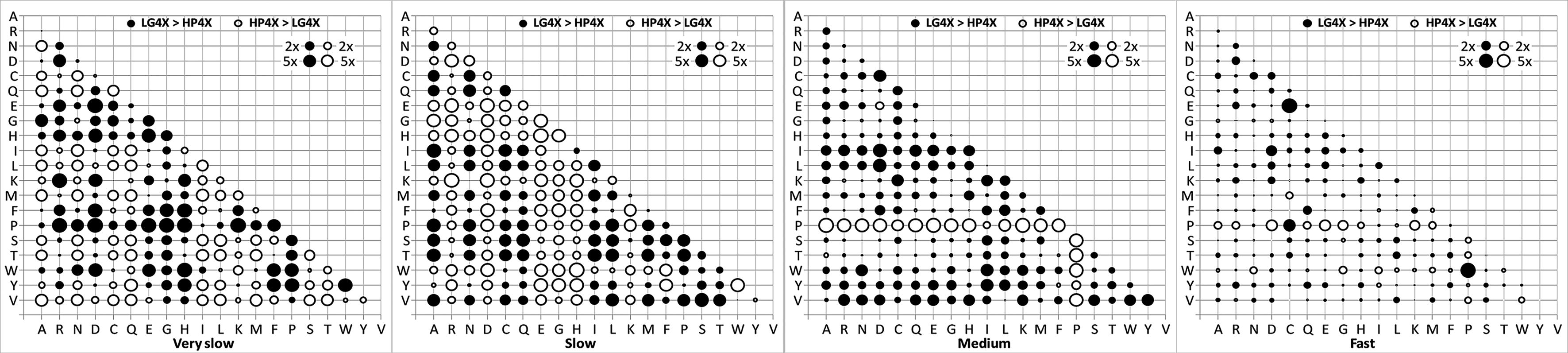
The relative difference between exchangeability coefficient in matrices of HP4X and LG4X models. Notations: 2x (5x) indicates that the exchangeability coefficient between two models is at least two times (five times) difference.
We compared the performance of four-matrix models (i.e., LG4M, LG4X, HP4M, HP4X) and general single-matrix models LG (Le and Gascuel, 2008) and Q.pfam (Minh et al., 2021) in inferring maximum likelihood trees on 300 HSSP testing alignments and 84 TreeBASE benchmark alignments (Sanderson et al., 1994) using the AIC criteria (see Table 2). The re-estimated models HP4M and HP4X had slightly better AIC values than their corresponding models LG4M and LG4X. Specifically, HP4M was better than LG4M on 160 (53%) HSSP alignments and 63 (75%) TreeBASE alignments. Similarly, HP4X was better than LG4X on 152 (50.6%) HSSP alignments; and 48 (57.1%) TreeBASE alignments. The multi-matrix models HP4M and HP4X were much better than the single-matrix models LG and Q.pfam. For example, HP4X was better than LG on 299/300 HSSP testing alignments. The results confirm that the multi-matrix models outperform the single-matrix models in building maximum likelihood trees.
Model Comparison on 300 HSSP and 84 TreeBASE Testing Alignments. #M1>M2: The Number of Alignments Where M1 Has Better AIC Value than M2
The QMix program as well as re-estimated models HP4X and HP4M are freely available at https://github.com/tinhnh2/Qmix. QMix can estimate a k-matrix mixture model from a set of protein alignments on a Linux computer. All training protein alignments are contained in one folder. The estimation process can be accomplished by one command
to execute the QMix program with the following parameters:
rate_model M: The type of the mixture model, i.e., M for the discrete gamma distribution and X for the distribution-free scheme ncat 4: The number of rate categories, e.g., 4 init_model LG: The initial replacement matrix, e.g., LG corr_threshold 0.99: The Pearson correlation used to stop the estimation process, e.g., 0.99 nthread 18: The number of computing threads, e.g., 18 threads data hssp1471: The full path to a folder of training alignments, e.g., hssp1471
The resulting mixture model contains k matrices named from Q.1 (the slowest rate) to Q.K (the fastest rate) created at the running folder. The resulting k-matrix models can be used to construct maximum likelihood trees by IQ-TREE2 software (i.e., iqtree2 -s alignment.phy -m “MIX{Q.1,Q.2,Q.3,Q.4}”). We note that QMix is written in Python; however, it can be converted to C++ to be incorporated into other more complex software such as IQ-TREE.
We presented the QMix program for estimating k-matrix mixture models using the maximum likelihood approach. The rate heterogeneity among sites can be modeled using either the discrete gamma rate distribution or the distribution-free scheme. Experiments on both the HSSP and TreeBASE alignments showed that the QMix program was able to estimate reliable and robust 4-matrix mixture models.
The QMix program takes a set of alignments and automatically estimates a k-matrix mixture model from the training alignments in an acceptable time, e.g., about ten hours to estimate four-matrix mixture models from 200 HSSP alignments. It is easy to use and applicable for researchers to estimate k-matrix mixture models from their datasets.
In summary, QMix improves the workflow used to estimate LG4M and LG4X by employing IQ-TREE2 and QMaker instead of PhyML and XRATE to accurately and rapidly estimate the parameters of models. It also supports multi-threading computing to efficiently estimate models from thousands of alignments. QMix can estimate mixture models with different number of matrices.
Footnotes
AUTHORS’ CONTRIBUTIONS
N.H.T.: Methodology (equal); Software (lead); Formal analysis (lead); Writing—original draft (lead); Visualization (equal). C.C.D.: Conceptualization (equal); Methodology (equal); Writing—review and editing (equal); Visualization (equal). L.S.V.: Conceptualization (equal); Methodology (equal); Writing—review and editing (lead); Supervision (lead).
AUTHOR DISCLOSURE STATEMENT
The authors declare that they have no conflicting financial interests.
FUNDING INFORMATION
No funding was received for this article.