
Abstract
Metagenomic Hi-C (metaHi-C) has shown remarkable potential for retrieving high-quality metagenome-assembled genomes from complex microbial communities. Nevertheless, existing metaHi-C-based contig binning methods solely rely on Hi-C interactions between contigs, disregarding crucial biological information such as the presence of single-copy marker genes. To overcome this limitation, we introduce ImputeCC, an integrative contig binning tool optimized for metaHi-C datasets. ImputeCC integrates both Hi-C interactions and the discriminative power of single-copy marker genes to group marker-gene-containing contigs into preliminary bins. It also introduces a novel constrained random walk with restart algorithm to enhance Hi-C connectivity among contigs. Comprehensive assessments using both mock and real metaHi-C datasets from diverse environments demonstrate that ImputeCC consistently outperforms other Hi-C-based contig binning tools. A genus-level analysis of the sheep gut microbiota reconstructed by ImputeCC underlines its capability to recover key species from dominant genera and identify previously unknown genera.
INTRODUCTION
Metagenomics is revolutionizing microbial ecology by enabling the exploration of complex microbial communities in diverse environments without the need for traditional microbial isolation or cultivation (Handelsman, 2004; Hugenholtz and Tyson, 2008; Simon and Daniel, 2011; Streit and Schmitz, 2004). The recent combination of Hi-C sequencing with whole metagenomic shotgun sequencing leads to the development of the metagenomic Hi-C (metaHi-C) technique, which has provided novel perspectives on species diversity and the interactions among microorganisms within a single microbial sample (Beitel et al., 2014; Burton et al., 2014; Du et al., 2023; Marbouty et al., 2014; 2021; Press et al., 2017; Yaffe and Relman, 2020). In metaHi-C experiments, shotgun sequencing extracts genomic fragments from a microbial sample, while Hi-C sequencing conducted on the same microbial sample generates DNA-DNA proximity ligations within the same cells, resulting in millions of paired-end Hi-C short reads. These fragmented shotgun reads are assembled into longer contigs, forming the basis for aligning paired-end Hi-C reads. MetaHi-C contacts, representing the number of Hi-C read pairs linking contig pairs, reveal contig relationships based on physical proximity within the microbial community. Depending on whether the shotgun libraries in metaHi-C experiments are constructed using second-generation or third-generation sequencing technologies, metaHi-C experiments can be classified into either short-read or long-read metaHi-C datasets, respectively.
Considering contigs originating from the same genome exhibit enriched Hi-C contact frequencies relative to those derived from distinct genomes, the process of Hi-C-based binning emerges and aims at grouping fragmented contigs into metagenome-assembled genomes (MAGs) (Hugerth et al., 2015) by leveraging Hi-C contacts between contigs (Baudry et al., 2019; DeMaere and Darling, 2019; Du and Sun, 2022; 2023). The resulting MAG collections serve as fundamental prerequisites for downstream analyses, such as the elucidation of the metabolic potentials and functional roles of diverse microorganisms, as well as the exploration of virus-host interactions (Chen et al., 2021; Gounot et al., 2022; Kent et al., 2020; Stalder et al., 2019). Various Hi-C-based contig binning methods have been developed, including HiCBin (Du and Sun, 2022), MetaTOR (Baudry et al., 2019), bin3C (DeMaere and Darling, 2019), and the MetaCC binning module (referred to as MetaCC) (Du and Sun, 2023). Compared to conventional shotgun-based binning tools reliant on sequence composition and contig coverage for contig clustering, Hi-C-based binning methods demonstrate their superior ability in MAG recovery using only one single sample (Du and Sun, 2022; Press et al., 2017).
However, existing Hi-C-based binning methods rely solely on Hi-C interactions for contig grouping, overlooking valuable biological information encapsulated within single-copy marker genes. These genes, present as single copies in the vast majority of genomes (Albertsen et al., 2013), hold the great potential to discriminate between contigs originating from distinct species when shared among them. This omission underscores a critical gap in current approaches, leaving ample room for enhancement and improved analyses. In response, we introduce ImputeCC, an integrative binning tool designed for metaHi-C datasets. ImputeCC manages to harness the comprehensive insights offered by both Hi-C interactions and single-copy marker genes to optimize the contig binning process. We conducted a comprehensive validation of ImputeCC’s performance using a combination of mock and real metaHi-C datasets. In the mock datasets, we demonstrated the effectiveness of our constrained random walk with restart (CRWR) imputation, showcasing its utility and necessity in improving the preclustering of marker-gene-containing contigs. Subsequently, we evaluated ImputeCC’s performance against other publicly-available Hi-C-based binning tools using four real metaHi-C datasets sourced from diverse environments, including the human gut (Press et al., 2017), wastewater (Stalder et al., 2019), cow rumen (Bickhart et al., 2019), and sheep gut (Bickhart et al., 2022). ImputeCC’s standout performance was particularly evident in the challenging sheep gut environment. In this complex setting, ImputeCC successfully retrieved an impressive total of 408 high-quality and 885 medium-quality MAGs, as assessed by the latest CheckM2 (Chklovski et al., 2023). To the best of our knowledge, this represents the largest number of reference-quality MAGs reported from a single sample. Moreover, we delved into the taxonomic diversity of the captured species in microbial samples by annotating high-quality MAGs generated by various binning methods using GTDB-TK (Chaumeil et al., 2022). ImputeCC consistently demonstrated a significantly broader taxonomic diversity at the species level across all datasets, emphasizing its ability to capture a broader range of microbial taxa. Further downstream ImputeCC’s genus-level analysis of the sheep gut microbiota revealed ability of ImputeCC to recover essential species from dominant genera such as Bacteroides, showed its potential to detect previously unrecognized genera, and unveiled other high-quality MAGs within the Alistipes genus that warrant further experimental investigation to elucidate their characteristics and roles within this ecosystem.
METHODS
Datasets
Mock metaHi-C datasets
The mock community sequencing data were downloaded from the European Nucleotide Archive under project ID PRJEB52977 (Meslier et al., 2022). The mock community comprises 71 strains representing 69 distinct species and underwent comprehensive sequencing using the Illumina HiSeq 3000, ONT MinION R9, and PacBio Sequel II platforms, generating three different shotgun libraries. The specific accession numbers and sizes of these three shotgun libraries are shown in Supplementary Table S1. After filtering the incomplete reference genomes (Supplementary Data S1), we obtained reference genomes of 66 distinct species for the following experiments. The abundances of all species were available from the supplementary data of (Meslier et al., 2022). Since the original dataset lacked Hi-C sequencing reads, we employed sim3C (v0.2) (DeMaere and Darling, 2018) to simulate metagenomic Hi-C reads based on the 66 reference genomes and their known abundances in the mock community, utilizing parameters ‘-n 10000000 -l 150 -e MluCI -e Sau3AI -m hic –insert-sd 20 –insert-mean 350 –insert-min 150 –linear –simple-reads’. Subsequently, we combined the same simulated Hi-C library with the three shotgun libraries, respectively, to construct three mock metaHi-C datasets. These mock Hi-C datasets were named according to the shotgun library incorporated in the mock dataset, resulting in the ‘mock Illumina,’ ‘mock PacBio,’ and ‘mock Nanopore’ metaHi-C datasets. Each mock dataset comprised real shotgun reads sequenced from a known mock community, along with simulated Hi-C reads.
Real metaHi-C datasets
Four publicly-available real metaHi-C datasets were utilized in this study, comprising two short-read metaHi-C datasets and two long-read metaHi-C datasets. The specific sizes of the raw datasets are detailed in Supplementary Table S2.
The two short-read metaHi-C datasets were derived from the human gut (BioProject: PRJNA413092) (Press et al., 2017) and wastewater (BioProject: PRJNA506462) (Stalder et al., 2019) samples. Each short-read metaHi-C dataset consisted of both shotgun and Hi-C libraries originating from the same sample source. The construction of Hi-C sequencing libraries involved the use of restriction endonucleases Sau3AI and MluCI. Sequencing of both the shotgun and Hi-C libraries was carried out on Illumina platforms, producing 150-base pair reads. The two long-read metaHi-C datasets were obtained from cow rumen (BioProject: PRJNA507739) (Bickhart et al., 2019) and sheep gut (BioProject: PRJNA595610) (Bickhart et al., 2022) samples. The cow rumen long-read metaHi-C dataset comprised uncorrected PacBio long-read libraries and Hi-C libraries. The error-prone PacBio long reads were generated using both the PacBio RSII and PacBio Sequel platforms. Hi-C libraries for this dataset were prepared using the Sau3AI and MluCI restriction enzymes and subsequently sequenced on an Illumina HiSeq 2000, producing 80-base pair reads. The sheep gut long-read metaHi-C dataset consisted of PacBio circular consensus sequencing (CCS) long-read libraries and Hi-C sequencing libraries. The PacBio CCS long reads, characterized by high accuracy with average Q scores exceeding 20, were referred to as HiFi reads. Distinct Hi-C libraries for the sheep gut long-read metaHi-C dataset were generated using the Sau3AI and MluCI restriction enzymes and sequenced at a length of 150 base pairs.
Data preprocessing
We first conduct essential read cleaning procedures using ‘bbduk’ from the BBTools suite (v37.25) (Bushnell, 2014) to address issues such as adaptor sequences, low-quality reads, and PCR duplication (Supplementary Data S2). For each metaHi-C dataset, reads from the shotgun library are assembled into longer contigs (Supplementary Data S3). After assembly, processed paired-end Hi-C reads are aligned to these contigs using BWA-MEM (v0.7.17) (Li, 2013) with the ‘−5SP’ parameter to prioritize the alignment with the lowest read coordinate as the primary alignment. Subsequent alignment filtering steps include the removal of unmapped reads, secondary and supplementary alignments, and alignments with low quality (nucleotide match length <30 or mapping score <30). We count Hi-C read pairs aligned to two contigs as raw Hi-C contacts between contigs and those contigs with fewer than two Hi-C contacts are excluded. Raw Hi-C contacts are normalized by NormCC (Du and Sun, 2023) with default parameters to eliminate the systematic biases derived from the number of restriction sites, contig length, and coverage.
The framework of ImputeCC binning
Detect assembled contigs with single-copy marker genes
Similar to MaxBin (Wu et al., 2014), we identify single-copy marker genes, which are genes typically found as single copies in the majority of genomes (Albertsen et al., 2013) within the assembled contigs. We accomplish this by employing FragGeneScan (Rho et al., 2010) and HMMER (v3.3.2) (Finn et al., 2011) (Supplementary Data S4).
Impute the metagenomic Hi-C contact matrix for contigs containing marker genes
The effective preclustering of contigs with single-copy marker genes partially depends on the expectation that marker-gene-containing contigs can be reliably linked through robust Hi-C interactions if they come from the same genome. However, this expectation encounters a practical limitation attributed to the localized characteristics of proximity ligations, which implies that even when two contigs share the same genomic origin, they may fail to establish Hi-C contacts if they are not in close spatial proximity within the cell, thereby contributing to the sparsity of the metagenomic Hi-C contact matrix (Du et al., 2022). To facilitate improved connections among marker-gene-containing contigs originating from the same genome through Hi-C interactions, we design a metagenomic Hi-C contact matrix imputation method. This involves employing a CRWR technique to amplify the within-cell Hi-C signals specially for marker-gene-containing contigs. Specifically, we define m and n as the number of contigs containing single-copy marker genes and the total number of assembled contigs, respectively. Let H denote the NormCC-normlized Hi-C contact matrix, where the entry
We use
To avoid the imputed matrix becoming too dense, we only retain the largest
Let Early stop if
Let
Leveraging the imputed Hi-C matrix Q as well as the characteristics of single-copy marker genes, we would like to accurately precluster contigs with marker genes as preliminary bins. Specifically, we first sort all categories of detected marker genes by the number of contigs containing the marker genes. If several marker genes correspond to the same number of contigs, they are further sorted by the gene length. Then, we use a greedy strategy to iteratively construct the preliminary bins as follows:
Initialization: Choose all contigs from the first marker gene and initialize preliminary bin set, denoted by Iteration: In the k-th iteration, we select all contigs containing the k-th marker gene and only handle contigs that have not been assigned to any preliminary bins in Repeat the iteration step until all marker genes are processed.
We apply the Leiden community detection algorithm (Traag et al., 2019) to the NormCC-normalized Hi-C contact matrix H to cluster all assembled contigs, using the preliminary bin set as an initial framework. The Leiden algorithm iteratively merges and refines communities to maximize modularity, a metric that quantifies the partitioning quality. To incorporate preliminary bin information, we initialize contig memberships based on preliminary bins, ensuring that contigs from the same preliminary bin are placed within the same community, while contigs not associated with any preliminary bins are initially assigned to individual communities. Throughout the Leiden iterations, these assignments for contigs from preliminary bins remain fixed. Consequently, contigs from the same preliminary bin coalesce into the same cluster, while those from different preliminary bins form distinct clusters after the Leiden clustering.
Moreover, since the Leiden algorithm is modularity-based, we select a flexible modularity function based on the Reichardt and Bornholdt’s Potts model (Reichardt and Bornholdt, 2006). Notably, the resolution parameter r in the modularity function (Supplementary Data S5) is a hyper-parameter that determines the relative importance assigned to the configuration null part compared to the links within the communities. To ascertain the optimal resolution parameter, we conduct parallel executions of the Leiden algorithm using various resolution values and automatically select the most favorable outcome. Specifically, we identify lineage-specific genes, which act as indicators of genome quality, through the application of the CheckM (v1.1.3) (Parks et al., 2015) function ‘checkm analyze’. Consequently, for any given contig bin, we employ the same evaluation strategy as CheckM to efficiently estimate its precision and recall (Supplementary Data S6). Subsequently, for each resolution parameter value, we count the number of genomic bins with precision exceeding 95% and recall surpassing 90%, 70%, and 50%, respectively. Finally, we automatically select the resolution value that maximizes the sum of three count numbers as the optimal choice.
Integrative strategy to obtain the final bins
It is essential to acknowledge that the preliminary bins may not be entirely accurate. This can occur, for instance, in cases where genome coverage is insufficient or marker genes are fragmented into several pieces. Furthermore, our clustering strategy in Subsection 2.3.4 may exacerbate these mis-binning arising from the preliminary bin assignments. Consequently, it is still meaningful to apply the Leiden algorithm to cluster contigs independently, without relying on the preliminary bin information. The selection of the resolution parameter follows the same methodology as previously described. We denote the resulting bin sets as
The MAG displaying the highest estimated quality across both bin sets is selected for further consideration. In situations where two or more MAGs exhibit identical estimated quality scores, ties are resolved by selecting the MAG with the greatest N50 statistic and bin size. Following the selection of a MAG, it is moved from the corresponding bin set to the final bin set, and any contigs belonging to the selected MAG are also removed from the other bin set, if present. This iterative procedure continues until the highest quality MAG identified falls below 10. Finally, we can obtain the final bin set through the integration.
For the mock metaHi-C datasets, where all species within the mock microbial community were known, the species identity of the assembled contigs could be determined (Supplementary Data S7). Then, we can define the completeness and contamination of each MAG recovered from the mock datasets. Specifically, for each MAG, we segregated the lengths of contigs according to their respective reference genomes and attributed the MAG to the reference genome with the largest cumulative contig length, denoted as
For the real metaHi-C datasets, since the actual genomes are unknown in real samples, we applied CheckM2 (Chklovski et al., 2023) to evaluate the completeness and contamination of retrieved MAGs. CheckM2 is an advanced machine learning-based method for assessing the quality of draft genomic bins, offering improved accuracy and computational speed compared to existing tools (Chklovski et al., 2023). Based on the CheckM2 assessments of completeness and contamination, we categorized the resolved MAGs from real metaHi-C datasets as high-quality if their completeness ≥90% and contamination ≤5%, while MAGs were designated as medium-quality if their completeness ≥50% and contamination ≤10%.
MAG analyses on real metaHi-C datasets
To assess the capacity of various binning methods in capturing taxonomic diversity within real metaHi-C datasets, we performed taxonomic annotation on all high-quality and medium-quality bins using GTDB-TK (v2.1.0, Release: R207 v2) (Chaumeil et al., 2022) with the function ‘classify_wf’ to extract the taxonomic information of the MAGs recovered by different binning methods.
Furthermore, to identify overlapping high-quality bins retrieved from the sheep gut long-read metaHi-C dataset between ImputeCC binning and other Hi-C-based binning approaches, we utilized Mash (v2.2) (Ondov et al., 2016) with 10,000 sketches per bin to calculate the Mash distance between high-quality bins from different bin sets. Bins with a Mash distance below 0.01 were considered MAGs originating from the same genome.
Other binners used in benchmarking
All binners used for comparison, i.e., VAMB (v3.0.3) (Nissen et al., 2021), HiCBin (v1.1.0) (Du and Sun, 2022), MetaTOR (v1.1.4) (Baudry et al., 2019), bin3C (v0.1.1) (DeMaere and Darling, 2019), and MetaCC (v1.1.0) (Du and Sun, 2023) were executed with default parameters on all mock and real metaHi-C datasets.
RESULTS
Overview of ImputeCC
ImputeCC is an integrative Hi-C-based binner that leverages the combined power of Hi-C interactions and single-copy marker genes in the contig binning process. Figure 1 shows the outline of ImputeCC. The core concept of ImputeCC involves the preclustering of marker-gene-containing contigs guided by two fundamental principles: I) Contigs sharing the same single-copy marker gene originate from distinct species with high probability; II) Contigs without overlapping single-copy marker genes are likely from the same genome when connected by robust Hi-C signals. To address the challenge that marker-gene-containing contigs from the same genome may not be effectively linked by Hi-C contacts due to the locality characteristics of proximity ligations, we design a new CRWR algorithm to impute the metaHi-C contact matrix before preclustering, with all random walks limited to start from marker-gene-containing contigs. Subsequently, by leveraging the imputed Hi-C matrix in conjunction with the aforementioned principles, ImputeCC can accurately precluster contigs with single-copy marker genes, establishing them as preliminary bins. Finally, the tool applies Leiden clustering (Traag et al., 2019) to group all assembled contigs, utilizing the information from preliminary bins to optimize the binning process.
ImputeCC achieved accurate preclustering for contigs containing Single-Copy marker genes
Since ImputeCC relies on the information provided by preliminary bins for final contig clustering, the quality of these preliminary bins, as established during the preclustering step, holds a pivotal role in affecting the final binning results of ImputeCC. Since the ground truth of all contigs from the mock metaHi-C datasets were known, we could leverage the mock datasets to assess the quality of the preclustering of preliminary bins. Specifically, we calculated the Adjusted Rand Index (ARI) clustering evaluation metric (Supplementary Data S8) for preliminary bins derived from the mock Illumina, Nanopore, and PacBio datasets (see Subsection 2.1.1), resulting in values of 0.976, 0.975, and 0.988, respectively (Fig. 2a). These values indicated that ImputeCC could accomplish precise preclustering for contigs with single-copy marker genes. Furthermore, we performed preclustering directly using NormCC-normalized Hi-C contacts, omitting the imputation step. In this context, the ARI values for preliminary bins derived from the three mock datasets were decreased to 0.783, 0.903, and 0.775, respectively (Fig. 2a), underscoring the significant enhancement in the construction of preliminary bins achieved through our CRWR imputation.
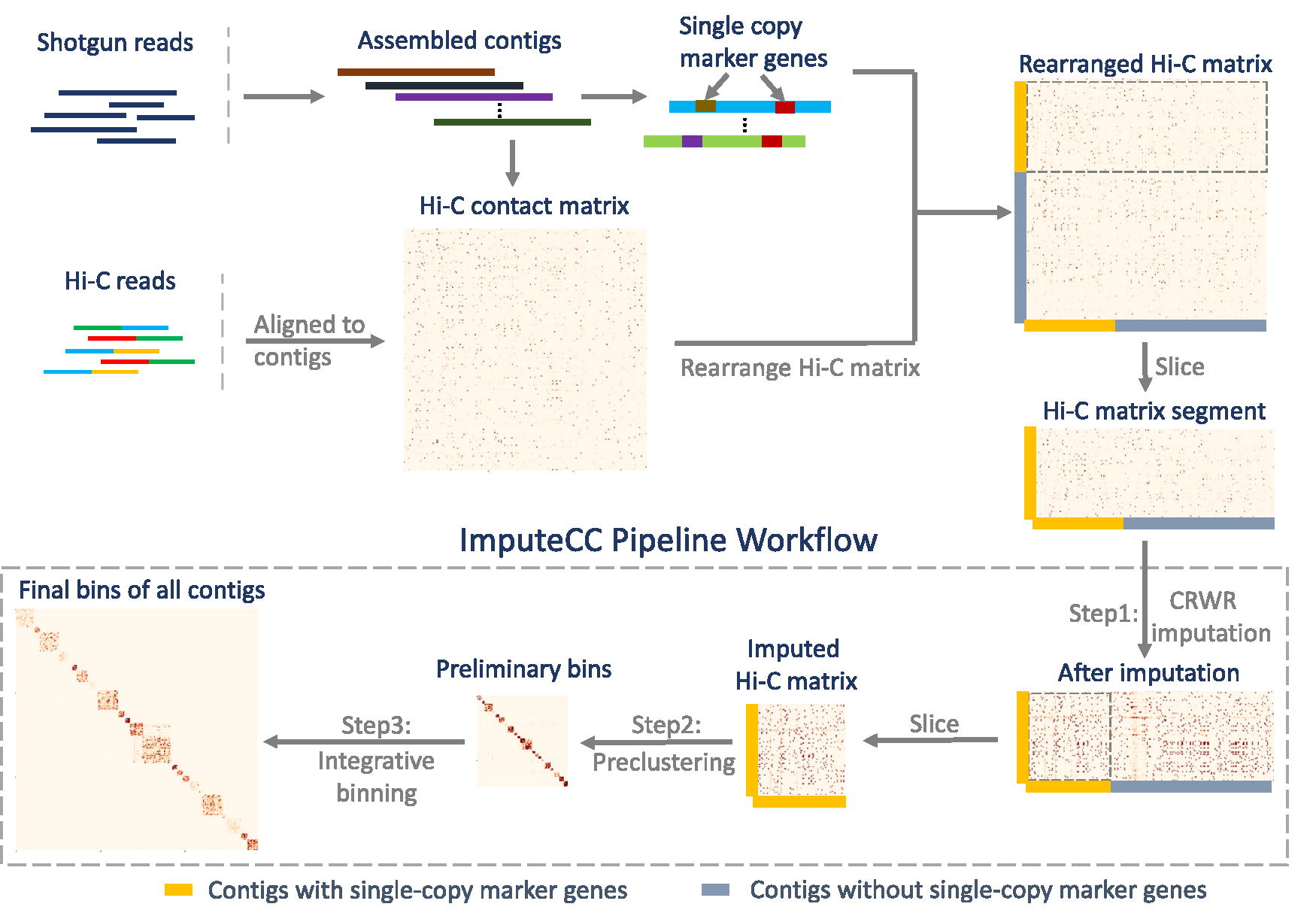
Overview of the ImputeCC. Given an input of the metagenomic Hi-C contact matrix and contigs containing single-copy marker genes, ImputeCC initiates the imputation of the metaHi-C contact matrix using a new CRWR algorithm, specifically limiting random walks to originate from contigs with marker genes. Subsequently, ImputeCC segregates and retains the imputed contact matrix exclusively for marker-gene-containing contigs, using it in conjunction with the characteristics of single-copy marker genes to effectively precluster these contigs as preliminary bins. Finally, the Leiden clustering method is applied by ImputeCC to group all assembled contigs, with insights from the preliminary bins guiding the optimization of the binning process. CRWR, constrained random walk with restart; metaHi-C, Metagenomic Hi-C.
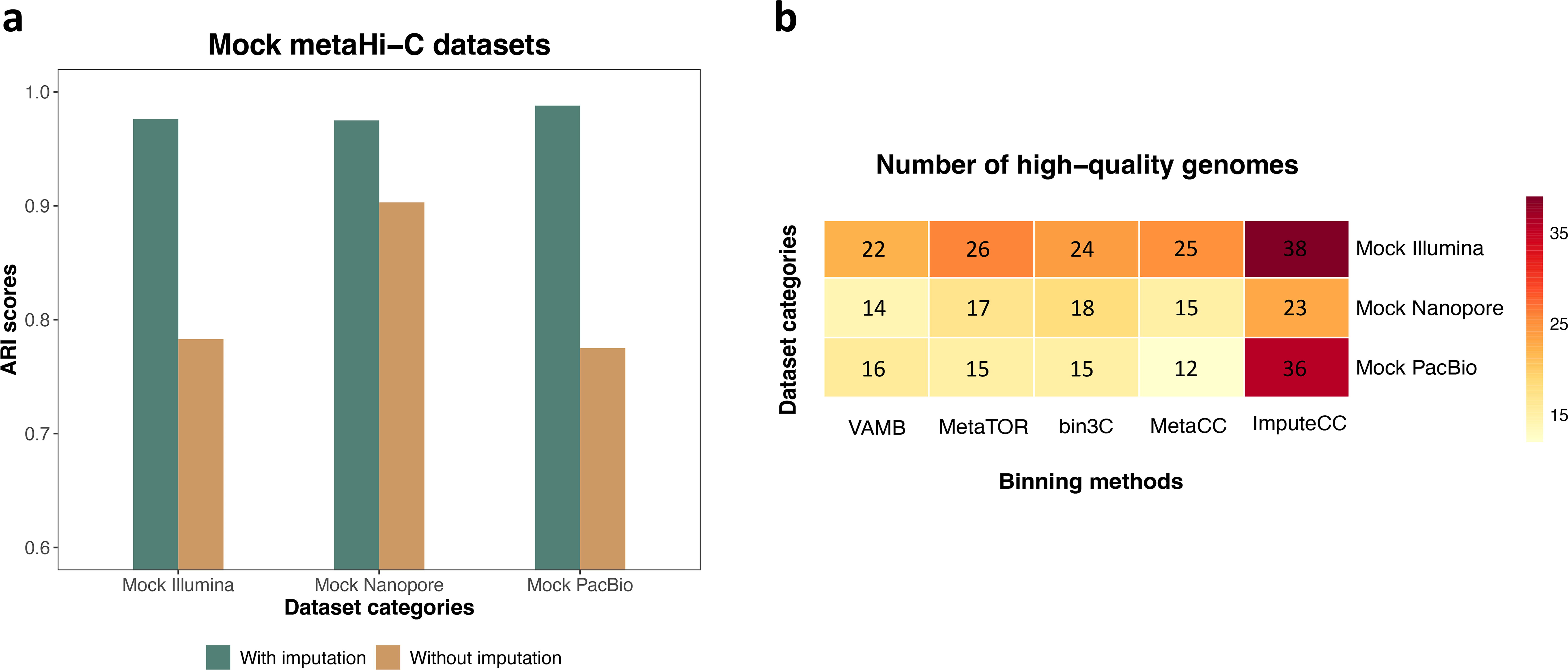
Benchmarking using the three mock metaHi-C datasets.
We first conducted a comparative evaluation of ImputeCC binning against VAMB (Nissen et al., 2021), MetaTOR (Baudry et al., 2019), bin3C (DeMaere and Darling, 2019), and the MetaCC binning module (referred to as MetaCC) (Du and Sun, 2023) using the three mock metaHi-C datasets. In addition to VAMB, a popular shotgun-based binning tool that utilizes sequence composition and coverage information, three other tools in consideration are Hi-C-based. It is important to note that another publicly available Hi-C-based binner HiCBin (Du and Sun, 2022) was excluded from the benchmarking study on the mock datasets due to its inability to converge when applied to the mock Nanopore and PacBio datasets. As shown in Figure 2b, ImputeCC demonstrated a remarkable ability to reconstruct a markedly larger number of high-quality genomes (completeness ≥90% and contamination ≤5%) across all the three mock datasets. Specifically, ImputeCC outperformed the second-highest result by 46.2%, 27.8%, and 125% in terms of high-quality genome reconstruction for the mock Illumina, Nanopore, and PacBio datasets, respectively. Notably, the number of mapped Hi-C read pairs for the mock Nanopore dataset was considerably lower in comparison to the mock Illumina and PacBio datasets (Supplementary Table S3), which can be attributed to the relatively higher error rate associated with Nanopore R9 long reads. This disparity in read mapping could be one of the contributing factors for ImputeCC retrieving a comparatively lower number of high-quality genomes from the mock Nanopore dataset. Finally, we evaluated ImputeCC’s stability against Hi-C sequencing depth by downsampling the Hi-C reads from 10 million to 5 million pairs in the mock datasets. The recovery of high-quality MAGs slightly declined from 38 to 36 in the Illumina dataset and from 23 to 21 in the Nanopore dataset, while the PacBio dataset consistently yielded 36 MAGs. These results highlighted ImputeCC’s resilience to reduced Hi-C read counts, ensuring its reliable performance in the mock metaHi-C datasets.
ImputeCC markedly outperformed existing binners on real metaHi-C datasets
To validate ImputeCC on real metaHi-C data, we applied it to two short-read and two long-read metaHi-C datasets from four different environments: human gut, wastewater, cow rumen, and sheep gut. Here, we compared ImputeCC to all four publicly-available Hi-C-based binners, namely HiCBin, MetaTOR, bin3C, and MetaCC, in addition to VAMB. Given the absence of reference genomes in real-world datasets, we utilized the CheckM2 (Chklovski et al., 2023) to evaluate the completeness and contamination of the recovered bins (see Subsection 2.4). In all cases, ImputeCC recovered more high-quality (completeness ≥90% and contamination ≤5%) and medium-quality (completeness ≥50% and contamination ≤10%) bins than the alternatives considered (Fig. 3a and b). Specifically, in the human gut and wastewater short-read metaHi-C datasets, ImputeCC reconstructed 66 and 75 high quality MAGs, outperforming the second-best binner with an increase of 11 (20%) and 10 (15.4%), respectively. For the cow rumen long-read metaHi-C dataset, though bin3C was able to retrieve an equivalent number of high-quality MAGs as ImputeCC, ImputeCC excelled by recovering 90.5% more medium-quality bins. The sheep gut long-read metaHi-C dataset, owing to its high complexity, posed a greater challenge. ImputeCC binning retrieved 408 high-quality MAGs, markedly outperforming VAMB, HiCBin, MetaTOR, bin3C, and MetaCC with an increase of 235 (135.8%), 321 (369%), 279 (216.3%), 160 (64.5%), and 82 (25.2%), respectively. ImputeCC was also able to recover 125.8%, 279.8%, 91.1%, 120.1% and 23.1% more medium-quality bins than VAMB, HiCBin, MetaTOR, bin3C, and MetaCC, respectively.
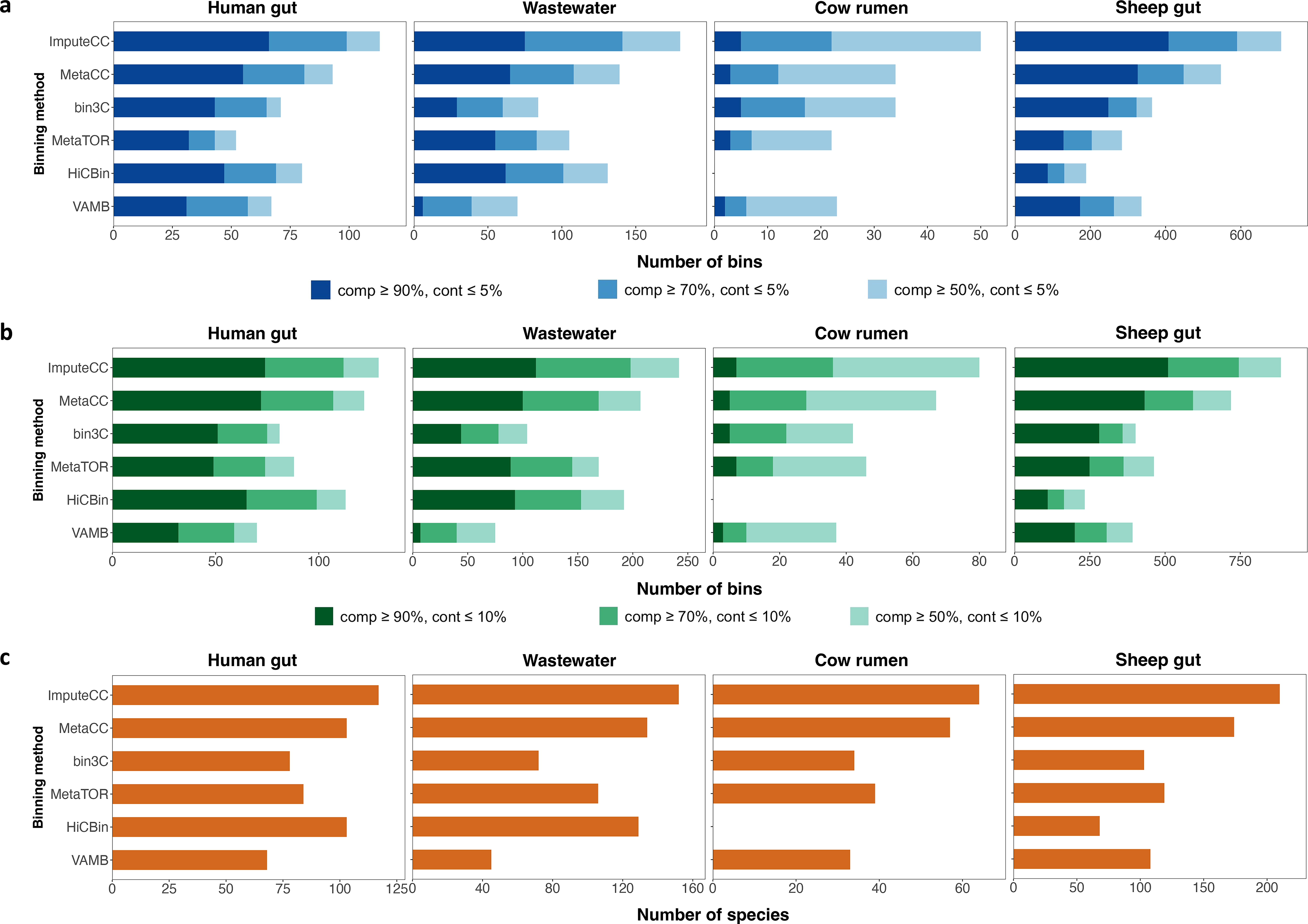
Benchmarking using the real human gut short-read, wastewater short-read, cow rumen long-read, and sheep gut long-read metaHi-C datasets.
Moreover, we explored the capability of different binners to capture the species diversity in microbial samples by annotating all medium-quality and high-quality bins generated by different binners on all real metaHi-C datasets using GTDB-TK (Chaumeil et al., 2022) (see Subsection 2.5). As shown in Figure 3c, medium-quality bins derived from ImputeCC represented a markedly larger taxonomic diversity at the species level on all datasets.
Finally, we conducted a detailed comparative analysis of the high-quality MAGs retrieved from the sheep gut long-read metaHi-C dataset. We employed Mash (Ondov et al., 2016) to identify cases where ImputeCC binning and three other Hi-C-based binning tools (MetaTOR, bin3C, and MetaCC) retrieved identical high-quality MAGs on the sheep gut long-read metaHi-C dataset (see Subsection 2.5). Notably, the majority of high-quality MAGs obtained through other Hi-C-based binning tools were also successfully recovered by ImputeCC (Fig. 4a). In contrast, ImputeCC binning went beyond by reconstructing a substantial number of high-quality MAGs that remained inaccessible to the other binning tools. Further annotation analyses of the high-quality MAGs demonstrated ImputeCC recovered more distinct taxa at various taxonomic levels compared to Hi-C-based alternatives, including bin3C, MetaTOR, and MetaCC (Fig. 4b).

Comparative analysis of high-quality MAGs retrieved from the sheep gut long-read metaHi-C dataset.
ImputeCC’s genus-level analysis, leveraging its retrieval of 408 high-quality MAGs, has unveiled significant insights into microbial composition of the sheep gut microbiota. Within this complex ecosystem, Bacteroides emerges as one of the dominant bacterial genera, well-recognized for its potential influence on the intestinal immune system (Routy et al., 2018; Yatsunenko et al., 2012). ImputeCC’s distinctive capabilities stood out as it successfully recovered two critical species from the Bacteroides genus, specifically Bacteroides uniformis and Bacteroides vulgatus, within the sheep gut environment. B. uniformis has garnered attention for its reported role in ameliorating immunological dysfunctions and metabolic disorders, often associated with intestinal dysbiosis (Gauffin Cano et al., 2012). In contrast, B. vulgatus assumes vital roles in reducing the production of gut microbial lipopolysaccharides and inhibiting atherosclerosis (Yoshida et al., 2018). Notably, among high-quality MAGs, while MetaCC managed to detect the presence of B. vulgatus, other binning tools failed to identify the genus Bacteroides from the sheep gut dataset. ImputeCC’s distinctive capability also emerged as it was the only method that could detect the Tidjanibacter genus, a relatively new and less-studied taxonomic group (Xie et al., 2020). This discovery creates opportunities for more research on this genus, offering the potential for exploring its ecological roles within the sheep gut environment. Within the Rikenellaceae family, ImputeCC’s analysis illuminated the prevalence and diversity of the Alistipes genus, which was predominantly found in the gastrointestinal tracts of the healthy human microbiome (Parker et al., 2020; Shkoporov et al., 2015). Specifically, ImputeCC retrieved 17 high-quality MAGs affiliated with Alistipes, compared to the 4, 3, and 9 high-quality MAGs recovered by MetaTOR, bin3C, and MetaCC, respectively. Among these 17 MAGs, Alistipes senegalensi emerged as a noteworthy species, recognized for its involvement in mannose fermentation (Mishra et al., 2012), suggesting a role of the members from the Alistipes genus within the sheep gastrointestinal tract’s intricate ecosystem. Furthermore, ImputeCC’s analysis unveiled five high-quality MAGs within the Alistipes genus that could not be annotated at the species level by GTDB-TK, suggesting the potential expansion of species diversity within the Alistipes genus. Additional experiments are necessary to gather further data on the phenotypic and physical characteristics of these uncultured members before their definitive identification can be achieved. In conclusion, all these findings underscore the unique efficacy of ImputeCC in advancing our understanding of microbial ecosystems by characterizing the sheep gut microbiota’s taxonomic composition and functional potential.
Running time analysis of the ImputeCC
On an Intel Xeon Processor E5-2665 with a clock speed of 2.40 GHz and 50 GB of allocated memory, the ImputeCC pipeline spent 64, 204, 25, and 2,115 min on the human gut short-read, wastewater short-read, cow rumen long-read, and sheep gut long-read metaHi-C datasets, respectively.
DISCUSSIONS
In this work, we developed ImputeCC, an integrative Hi-C-based contig binning methods. ImputeCC combines Hi-C interactions with the intrinsic discriminative potential of single-copy marker genes by preclustering marker-gene-containing contigs as preliminary bins. To enhance the Hi-C connectivity of marker-gene-containing contigs, ImputeCC introduces a CRWR approach to impute the metaHi-C contact matrix. Finally, ImputeCC employs Leiden clustering to group all assembled contigs, optimizing the binning process by leveraging information from the preliminary bins. Evaluations of ImputeCC using a wide range of diverse mock/real metaHi-C datasets have demonstrated its effectiveness for retrieving reference-quality MAGs and shown its potential to unravel the structure of microbial ecosystems and their resident microorganisms. Notably, we utilized CheckM2 in assessing the binning performance for the four real metaHi-C datasets. Although CheckM2 represents the most advanced software for evaluating bin quality in real metagenomic samples, it is essential to delve further into the accuracy of this machine-learning-based validation method in reflecting the true completeness and contamination levels of the recovered MAGs. Moreover, previous research has established the efficacy of Hi-C-based binning over shotgun-based approaches (DeMaere and Darling, 2019; Du and Sun, 2022). Accordingly, our benchmarking analyses focus on Hi-C-based methods, comparing ImputeCC with similar tools and including VAMB as a reference shotgun-based method.
ImputeCC offers several promising avenues for expansion. For instance, when dealing with large MAGs characterized by high abundances, there is potential in imputing normalized Hi-C contacts for contigs within these MAGs to facilitate the scaffolding process. Moreover, exploring imputation methods that consider additional information, such as the sequence composition of contigs, could yield improved imputation results.
Footnotes
AUTHORS’ CONTRIBUTIONS
Y.D. and F.S. conceived the ideas and designed the study. Y.D. implemented the methods, carried out the computational analyses, and drafted the article. Y.D. and W.Z. developed the software. All authors modified and finalized the paper.
AVAILABILITY OF DATA AND MATERIALS
The mock community sequencing data were downloaded from the European Nucleotide Archive under project ID PRJEB52977 (Meslier et al., 2022). The human gut short-read metaHi-C dataset used in this study is available under NCBI BioProject PRJNA413092 (Press et al., 2017). The wastewater short-read metaHi-C dataset is available under NCBI BioProject PRJNA506462 (Stalder et al., 2019). The cow rumen long-read metaHi-C dataset used in this study is available under NCBI BioProject PRJNA507739 (Bickhart et al., 2019). The sheep gut long-read metaHi-C dataset is available under NCBI BioProject PRJNA595610 (Bickhart et al., 2022). The ImputeCC software is freely available at ![]() .
.
CONSENT FOR PUBLICATION
All authors have approved the article for submission.
AUTHOR DISCLOSURE STATEMENT
The authors declare that they have no competing interests.
FUNDING INFORMATION
The research is partially funded by NSF grant
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.