
Abstract
Protein abnormalities disrupt various cellular and contribute to disease development. Identifying disease-associated proteins is crucial for precision medicine, but traditional methods are time-consuming and costly, necessitating computational approaches. Existing computational methods rely on manual feature engineering and fail to leverage deep features from amino acid sequences and protein structures. In this article, we propose Model for predicting protein–phenotype associations by Fusing multi-view Features (MFF-HPO), a model for predicting protein–phenotype associations by fusing multi-view features from amino acid sequences. First, we generate three-dimensional protein structure from amino acid sequence to derive contact graphs and secondary structures then integrate these with direct sequence encoding and physicochemical properties. Using a Graph Attention Network, we extract structural features from contact graphs, while deep neural networks capture global and local features from secondary structures, physicochemical properties, and sequence encoding. Finally, concatenated features are used to predict phenotype annotations. MFF-HPO outperforms state-of-the-art methods with a mean area under the precision-recall curve of 0.314 and a mean Fmax of 0.371. Ablation studies confirm that multi-view feature fusion enhances predictions, and case studies validate its practicality.
INTRODUCTION
Understanding how human proteins influence disease phenotypes is crucial for the development of precision medicine (Leopold and Loscalzo, 2018). To this end, Robinson et al. (2008) established the Human Phenotype Ontology (HPO), a standardized tool designed to describe phenotypic abnormalities and clinical features associated with human diseases. The advent of HPO has significantly enhanced our comprehension of the interplay between disease phenotypes and genes or proteins, facilitating disease identification and supporting medical research endeavors. Despite the identification of over 200 million proteins in protein databases such as Uniprot, a mere 4.35% have been explicitly linked to disease phenotypes (UniProt Consortium, 2023). This indicates that progress in human proteome research, studies on the connections between proteins and diseases are still in their infancy, with many associations between proteins and disease phenotypes yet to be discovered. The prohibitive costs associated with experimental exploration of these links highlight the critical need for computational strategies to unearth potential protein-disease phenotype associations.
Many computational methods have been proposed to predict associations between proteins and disease phenotypes. Researchers mainly focus on obtaining protein information from multiple perspectives, such as protein function, protein-protein interactions, and protein expression, aiming to accurately represent protein features (Liu, Mamitsuka, et al., 2022; Doğan, 2018; Liu, He, et al., 2022; Bi et al., 2023a). For instance, Liu, Mamitsuka, et al. (2022) utilized graph convolutional networks, aggregating phenotypic information from multiple protein interaction networks, to predict associations between proteins and disease phenotypes. Doğan (2018) identified potential protein-disease phenotype associations by analyzing the co-occurrence frequency of protein functions and disease phenotypes. These methods are based on the principle that “proteins that interact with each other tend to cause the same disease”. Moreover, the known functional information will enhance the protein feature representation. For example, Liu, He, et al. (2022) constructed protein attribute graphs using protein function and interaction information, and aggregated potential phenotypic information for each node through a variational graph autoencoder. Bi et al. (2023b) constructed an attribute graph including gene functions and protein-protein interactions (PPIs), obtaining feature representations of genes through a pre-trained model for downstream phenotype prediction task. The diverse sources of protein functions, interactions between proteins, and protein expression information provide rich data for protein feature representation (Liu et al., 2020). However, these methods rely on auxiliary information such as protein-protein interactions and protein functions limiting their applicabilities, as only a small fraction of proteins have such auxiliary information compared to the well-known amino acid sequences (Kulmanov and Hoehndorf, 2020).
The amino acid sequence is fundamental to a protein’s structure and functionality, playing a significant role in its physiological functions within organisms. Amino acid sequences of proteins are widely used in many research fields including protein function prediction (Gligorijević et al., 2021), interaction prediction (Soleymani et al., 2022), and drug target identification (Zhou et al., 2021). Through the analysis of amino acid sequence order, researchers utilizing deep learning have elucidated intrinsic biological properties, leading to the development of diverse methodologies (Jumper et al., 2021; Chowdhury et al., 2022; Gelman et al., 2021; Huang et al., 2021). These studies highlight that amino acid sequences encapsulate substantial biological insights, reflecting the diverse biological activities of proteins. However, despite the wealth of information encoded in amino acid sequences, predicting associations between proteins and phenotypes using these sequences still faces challenges. Previous efforts have often focused on amino acid composition (Gong et al., 2016; Zhang and Sagui, 2015; Almagro et al., 2017), overlooking critical biological insights such as the protein three-dimensional (3D) structures and the physicochemical characteristics of amino acids, which are crucial for determining protein behavior (Li et al., 2014; Dawson et al., 2017). The protein secondary structure is closely related to protein functions. Different structures endow proteins with distinct physical and chemical properties, which in turn influence various functional aspects such as protein-protein interactions and ligand binding (Kambouris et al., 2014). Additionally, physicochemical properties, such as polarity and acidity-alkalinity, play a significant role in protein folding, stability, and overall functionality (Song et al., 2024). Furthermore, the experimentally validated phenotype annotations for proteins remain sparse and exhibit a severe imbalance, further complicating the prediction of protein–phenotype associations.
To address these challenges, we introduce Model for predicting protein–phenotype associations by Fusing multi-view Features (MFF-HPO), a novel model designed to predict protein–phenotype associations by fusing multiple features extracted from amino acid sequences. MFF-HPO integrates protein features from three aspects: structure, physicochemical properties, and sequence composition, enabling accurate predictions of associations between proteins and phenotypes. Specifically, we first employ AlphaFold to generate 3D protein structures from amino acid sequences. Then, from the 3D structure, we construct the protein’s contact graph by calculating the amino acid distance and extract protein’s secondary structure. Following this, we apply one-hot encoding to represent the secondary structure, physicochemical properties, and amino acid composition of protein. Next, we leverage multi-head GAT to extract protein features based on the protein contact graph and design feature representation modules based on deep neural network from both global and local perspectives. Finally, we integrate these features and input them into a three-layer fully connected neural network to predict protein–phenotype associations. Experimental results demonstrate the effectiveness of our model in improving the prediction of protein–phenotype associations.
MATERIALS AND METHODS
Datasets
The amino acid sequences of proteins are obtained from the Uniprot database and filtered through Swiss-Prot to ensure reliability. The protein–phenotype associations are sourced from the HPO database. With the Gene-phenotype relationships from the HPO database released in October 2021, genes were mapped to proteins using the provided UniProt mapping tool. In cases where a gene maps to multiple proteins, all associations between these proteins and corresponding phenotypes are retained. Subsequently, the “true-path-rule”(Valentini, 2011) is applied to propagate proteins from child nodes to parent nodes in the obtained protein–phenotype associations. Following Liu’s method (Liu et al., 2020), phenotype terms with associated protein counts less than 11 are excluded to enhance the dataset reliability. Finally, 4,629 proteins, 4,575 HPO terms, and 680,660 relationships of proteins and HPOs are retained in the dataset.
The overview of the proposed method
In this article, we introduce the model MFF-HPO, which integrates the multiple protein features extracted from amino acid sequences to predict disease phenotypes. The overview of MFF-HPO is shown in Figure 1, comprising three steps: (A) encoding amino acid sequences from the perspectives of structure, physicochemical properties, and amino acid composition; (B) capturing structural features from protein structure graphs using GAT, and capturing global and local features from amino acid sequences using methods based on DCN and CNN respectively; (C) concatenating the multiple features and employing an MLP to predict the protein-HPO term associations.
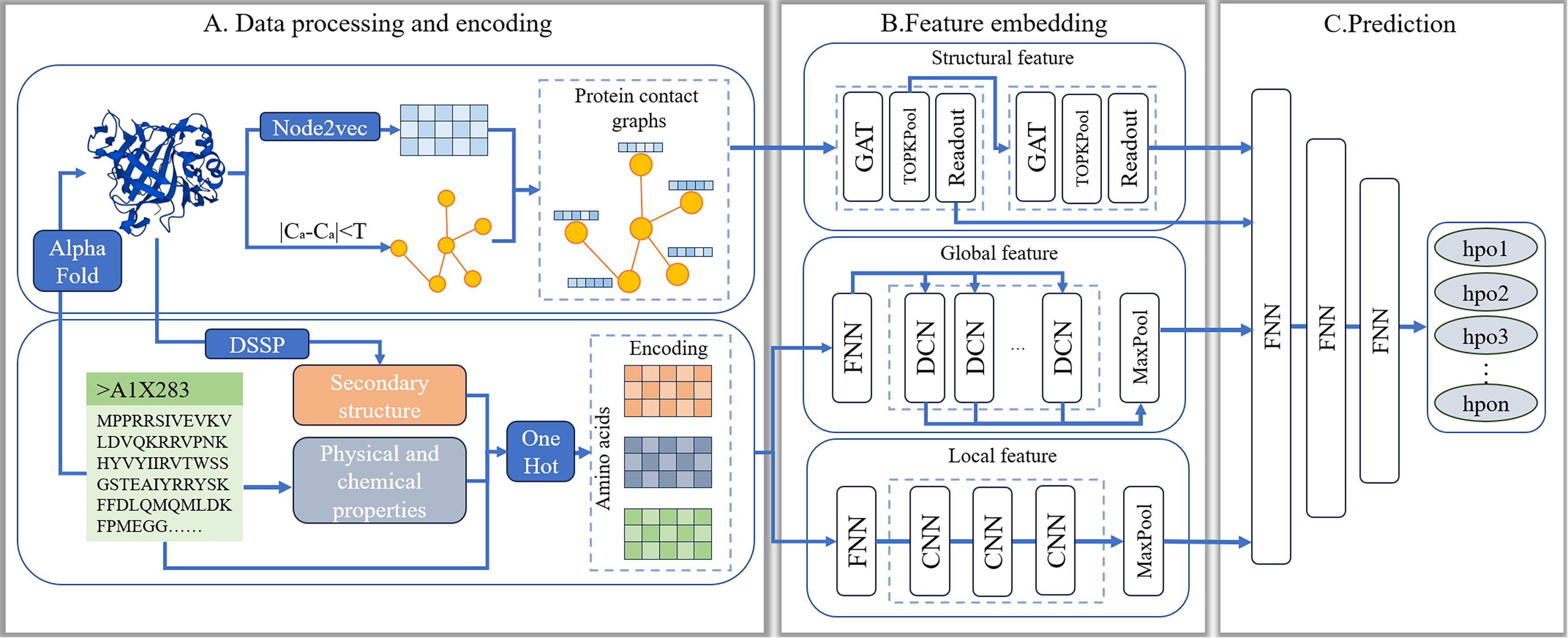
The overview of MFF-HPO.
Protein contact graph construction
The 3D structure of a protein has a significant influences its function. We use AlphaFold to obtain the 3D structure of proteins, which has been proven to be highly reliable. Following the method of Gligorijevic et al. (2021), we determine edges between amino acids based on the distances between their central carbon atoms. An edge is created if this distance is less than 10 Å, indicating direct interactions between residues. Then, Node2vec is applied to obtain the amino acids features from 3D structure of protein. The protein contact graph is constructed as
Amino acid sequence coding
In this study, we standardize the length of all amino acid sequences to 2,000, as more than 99% of sequences in UniProt are shorter than 2,001 (Kulmanov and Hoehndorf, 2020). During standardization, sequences longer than 2,000 amino acids are truncated to include only the initial 2,000 amino acids. Conversely, sequences shorter than 2,000 amino acids are extended by padding with ‘-’ to reach the standardized length. We focus solely on the 20 common amino acids, representing any non-standard residues and placeholders uniformly with the placeholder ‘-’.
Based on the standardized amino acid sequences, we perform one-hot encoding on the protein’s secondary structure, physicochemical properties, and sequence composition respectively. Firstly, we employ the DSSP (Zeng et al., 2020) to identify the secondary structures within protein 3D structures including 9 categories
Structural feature extraction
To capture effective information of structure from the constructed protein contact graph G, we construct a structural feature extraction module by integrating Multi-head Graph Attention Layers (MHGAT) with top-k pooling layers.
Following the idea of GAT, for the nodes i and j in graph G, the attention coefficient
Based on the calculated attention coefficient, the feature vector
To reduce computational complexity while retaining important information from the contact graph, we apply a top-k pooling operation to remove less important nodes in the graph after GAT. By setting a pooling rate k, the input contact graph G with N nodes is pooled into a subgraph
The function of proteins are influenced by the interactions of amino acids within their local environment. We utilize a convolutional neural network(CNN) composing three one-dimensional(1D) convolutional layers to extract multi-scale local features from the amino acid sequences. Specifically, the input amino acid sequence features
We employ a 3-layer CNN, with the kernel size set to [32, 64, 256] for each convolutional layer. Then, after a max pooling layer, the final local features vector
The global features of amino acid sequences can help understand the changes in proteins during evolution. We use dilated convolutional neural networks (DCN) to capture multi-scale global features from the protein’s amino acid sequence. Five DCNs with different dilation rates expand the receptive field. For the input sequence feature
In the prediction module, we use a three-layer fully connected neural network with a hidden layer of 512 to predict the associations between proteins and phenotypes. In practice, we first concatenate
In the last layer of the fully connected neural network, we use the Sigmoid function to control the prediction between 0 and 1. The predictive score for the input protein and HPO term j can be calculated as:
Experiment settings
In this study, we implement the MFF-HPO model using the Python and the PyTorch framework, on a computing environment equipped with V100-SXM2-32GB GPUs. To comprehensively evaluate the performance of MFF-HPO, we employ a fivefold cross-validation method in our experiments and use the Fmax score to assess the model’s performance (Bi et al., 2023). Specifically, we divided the dataset into five equal parts based on protein, using four parts as the training set and one part as the test set for each fold. Each train set contains 3,703 proteins and the all 4,575 HPO entries. Since the proteins used for training differ in each fold, the protein-HPO associations included also vary. In addition, given that the number of negative samples in the dataset significantly exceeds that of positive samples, this imbalance could bias the evaluation of model performance. Therefore, we also use area under the precision-recall curve (AUPR) to reflect the model’s prediction capability for positive samples.
Comparison with baseline methods
We compare MFF-HPO with five baseline methods, including Basic Local Alignment Search Tool (BLAST) (Tatusova and Madden, 1999), Naive (Clark and Radivojac, 2011), DeepFRI (Gligorijević et al., 2021), Deep_CNN_LSTM_GO (Elhaj-Abdou et al., 2021), and DeepGoPlus (Kulmanov and Hoehndorf, 2020) to demonstrate the method performance. The BLAST algorithm predicts phenotypes based on the similarity between amino acid sequences, while the Naive method relies on the frequency of phenotypes appearing in the database for prediction. Additionally, DeepFRI, DeepGoPlus, and Deep_CNN_LSTM_GO are three methods specifically designed for gene ontology term prediction, which are the tasks of the same type as phenotype prediction. These models utilize amino acid sequence information and employ various deep learning methods to accomplish the prediction task. To maintain fairness, we conduct the baselines using the setting described in their respective articles.
The experimental results demonstrate that our method MFF-HPO achieves the best performance on both metrics compared to all baseline methods, with a mean AUPR of 0.314 and a mean Fmax of 0.371, as shown in the Figure 2a. Among all baseline methods, DeepFRI achieved the second-best prediction performance with mean AUPR of 0.301 and mean Fmax of 0.365, following closely behind MFF-HPO. This may be attributed to the fact that this method also utilizes the 3D structure of amino acid sequences as protein features. The BLAST achieves the lowest result, indicating that it is difficult to effectively distinguish the function of proteins based solely on the similarity between amino acid sequences. Protein spatial structure and amino acid positions significantly influence protein function, highlighting the benefits of incorporating protein spatial structural information into phenotype prediction.
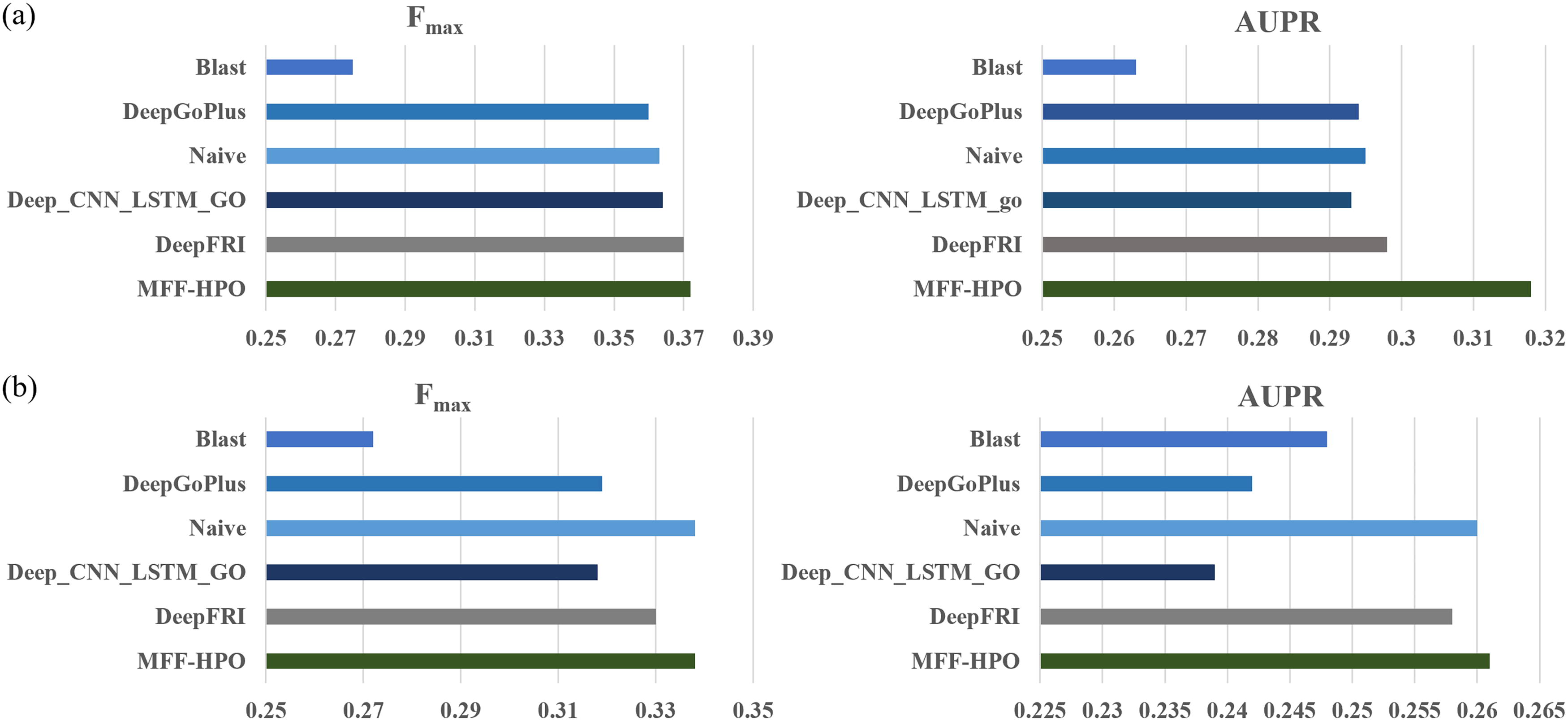
MFF-HPO experimental results.
To further validate the performance of the model, we train it using the 2021 version of the dataset and predicted the phenotype annotation of proteins added to the database from 2021 to 2023. As shown in Figure 2b, the predictive performance of all models decreases, which may be due to the lack of known phenotype annotations or sparse phenotype annotations for the newly added proteins. Our method continues to display the highest mean AUPR and mean Fmax values, proving its excellent robustness. Interestingly, the Naive method based on statistics also shows good performance in this scenario, even surpassing deep learning techniques such as DeepFRI, DeepGoPlus, and Deep_CNN_LSTM_GO. This result indicates that research on predicting protein-disease phenotype associations based on amino acid sequences is still in its early stage.
Prediction performance using different features
We conduct ablation experiments to demonstrate the rationality of the model design. We analyze the impact of different protein features on the model, by removing each protein feature and testing the predictive performance of the model, as shown in Figure 3a. By systematically removing different protein features from MFF-HPO, we observe varying degrees of decline in the model’s predictive performance, proving that each type of feature contributes to MFF-HPO’s accuracy. Particularly noteworthy is the significant decrease in MFF-HPO’s AUPR and Fmax scores following the removal of the physicochemical properties of amino acids, further validating the critical role of amino acid physicochemical characteristics in predicting disease phenotypes. The model’s performance changes the least after removing the one-hot encoding features of amino acids, indicating that, compared to other features, one-hot encoding provides relatively less effective information in the prediction process.

MFF-HPO experimental results.
The model can improve its performance in processing graph data by stacking multiple GAT layers. However, too many GAT layers also increase the risk of the oversmoothing problem. We vary the number of GAT layers in the MFF-HPO model to identify the optimal layer setting for the best predictive performance. As illustrated in Figure 3b, MFF-HPO achieves the best prediction results when the GAT layer count is set to 2. However, the predictive performance of MFF-HPO began to decline gradually when the number of GAT layers exceeded 2. This could be attributed to the structural module that integrates the output of each GAT layer into the final output. As the number of layers increases, the noise introduced by the GAT layers outweighs the valuable information they provide, leading to a decrease in the prediction performance of MFF-HPO.
Case study
Following the method of Liu et al. (2020), we conduct case study to validate the practicality of our model. Protein Q9C0G0 (Zinc finger protein 407) is a protein produced through the transcription and translation processes of the ZNF407 gene. Misexpression of this protein can lead to an autosomal recessive inherited cognitive disorder syndrome (Kambouris et al., 2014). This protein is a new added after 2020. In the temporal validation, we get the predicted HPO term list of it. Then, we rank the term list based on predicted scores. The top-5 terms can be validated in 2023 HPO dataset. We further search evidences in PubMed, shown in Table 1. All the top-5 predicted results are supported by corresponding literature evidence. This indicates that our model can achieve phenotype prediction for proteins without any prior knowledge and has good generalization performance.
Predicted Top-5 Phenotype Terms of Q9C0G0 and Evidences
Predicted Top-5 Phenotype Terms of Q9C0G0 and Evidences
HPO, Human Phenotype Ontology; Q9C0G0, Zinc finger protein 407.
Elucidating the associations between human proteins and disease phenotypes is crucial for the prevention, diagnosis, and treatment of diseases. This study introduces a prediction model MFF-HPO based on the fusion of multiple features from amino acid sequences, aiming at predicting associations between proteins and phenotypes. The proposed method represents protein features from different perspectives. Constructing protein contact graphs from amino acid 3D structures provides internal spatial information of the amino acid sequence. The protein’s secondary structure further enriches the structural information. By integrating the physicochemical properties of proteins and the direct encoding of sequences, the model captures global and local features of amino acid sequence. The fusion of multiple features enhances the prediction of phenotype annotations. Ablation experiments and case studies also confirm the validity of our proposed method, which can serve as a useful tool for clinical applications utilizing protein sequence information.
However, it should be noted that the HPO database has been enhanced with the advancement of clinical phenotype genomics. The HPO’s phenotype ontology follows a directed acyclic graph structure. In future research, incorporating the hierarchical structure of phenotypes will significantly advance the exploration of associations between proteins and phenotypes in the scientific field.
Footnotes
AUTHORS’ CONTRIBUTIONS
X.B. and Z.J.: Conceptualization, writing—original draft, review and editing. L.Z. and K.Z.: Project administration, resources (supporting role). G.Y. and Z.G.: Review the draft.
AUTHOR DISCLOSURE STATEMENT
The authors declare that they have no competing interests.
FUNDING INFORMATION
This work is supported by the Natural Science Foundation of Xinjiang Uygur Autonomous Region (Nos. 2024D01C126, 2022D01C427, and 2022D01C429), the National Natural Science Foundation of China (No. 62366052, No. 12061071), the Key R&D Program of Xinjiang Uygur Autonomous Region (No. 2022B03023, No. 2022B01046), and The 20th International Symposium on Bioinformatics Research and Application (ISBRA 2024).