
Abstract
CRISPR-Cas clinical trials have begun, offering a first glimpse at how DNA and RNA targeting could enable therapies for many genetic and epigenetic human diseases. The speedy progress of CRISPR-Cas from discovery and adoption to clinical use is built on decades of traditional gene therapy research and belies the multiple challenges that could derail the successful translation of these new modalities. Here, we review how CRISPR-Cas therapeutics are translated from technological systems to therapeutic modalities, paying particular attention to the therapeutic cascade from cargo to delivery vector, manufacturing, administration, pipelines, safety, and therapeutic target profiles. We also explore potential solutions to some of the obstacles facing successful CRISPR-Cas translation. We hope to illuminate how CRISPR-Cas is brought from the academic bench toward use in the clinic.
Introduction
Gene therapy holds the promise of treating disease-causing genetic, epigenetic, and transcriptomic aberrations. The first couple of decades of research and development in this field were marked by eager exploration and some devastating setbacks. 1 Even so, persistence has begun to pay off with the first generation of approved gene therapies that augment missing or defective genes in conditions such as transfusion-dependent β-thalassemia, spinal muscular atrophy (SMA), and inherited retinal disease.2–4
Fortuitously, the long-awaited emergence of these genetic therapies coincides with the recent advent of genome and transcriptome engineering tools based on the CRISPR-Cas RNA-guided nuclease systems, which have transformed our ability to precisely manipulate nucleic acids. CRISPR-Cas has expanded our genome editing toolkit, enabling precision alteration, replacement, and regulation of genes to therapeutically target disease states and potentially confer disease resistance. Beyond genetic diseases, CRISPR-Cas editing therapies have been used to target acquired diseases such as cancer, cardiovascular disease, infectious disease, and chronic ailments such as Alzheimer's disease (AD) in animal models.5–8 The nexus of these two fields has created extraordinary excitement for the potential of CRISPR-based gene therapies to revolutionize the treatment of many of the most important ailments currently impacting human health.
Although that potential has yet to be fully realized, preclinical studies of CRISPR-based therapies have exploded in recent years. Pioneering CRISPR-Cas gene therapy clinical trials have begun treating some monogenic diseases and severe cancers. Still, the broad application of CRISPR-Cas therapies has a long way to go and hinges on multiple factors. These factors include long-term studies to demonstrate safety without off-target activity; advancement in precise and effective delivery systems; immune system interactions; efficient and cheap manufacturing; and effective regulation to democratize access to these therapies.
In this review, we cover the steps that are necessary to take CRISPR-based gene therapies from concept through drug design and preclinical testing to clinical trials. This process is complex and may be iterative with many decision points and important milestones along the way. We then focus on the most critical challenges facing the development of these therapies, especially in delivery, administration, safety, preclinical study design, drug manufacturing, and economic and regulatory issues. We describe the obstacles they present and how studies to date have approached these issues. We hope to offer both a window into the current state of the field and a map to orient thought and resources toward solving the most significant challenges that may impede the realization of the potential of CRISPR-based gene therapy.
Therapeutic Development Cascade
Development of a gene therapeutic is an enormously complex endeavor,5,9 weighing strategies incorporating unmet medical need, editing approach, needed repair percentages, availability of disease models that faithfully recapitulate human biology, and delivery considerations among a multitude of aspects that are crucial to creating a safe and efficacious medicine. Correspondingly, the path toward enabling a new therapy follows a multifaceted development cascade (Fig. 1).
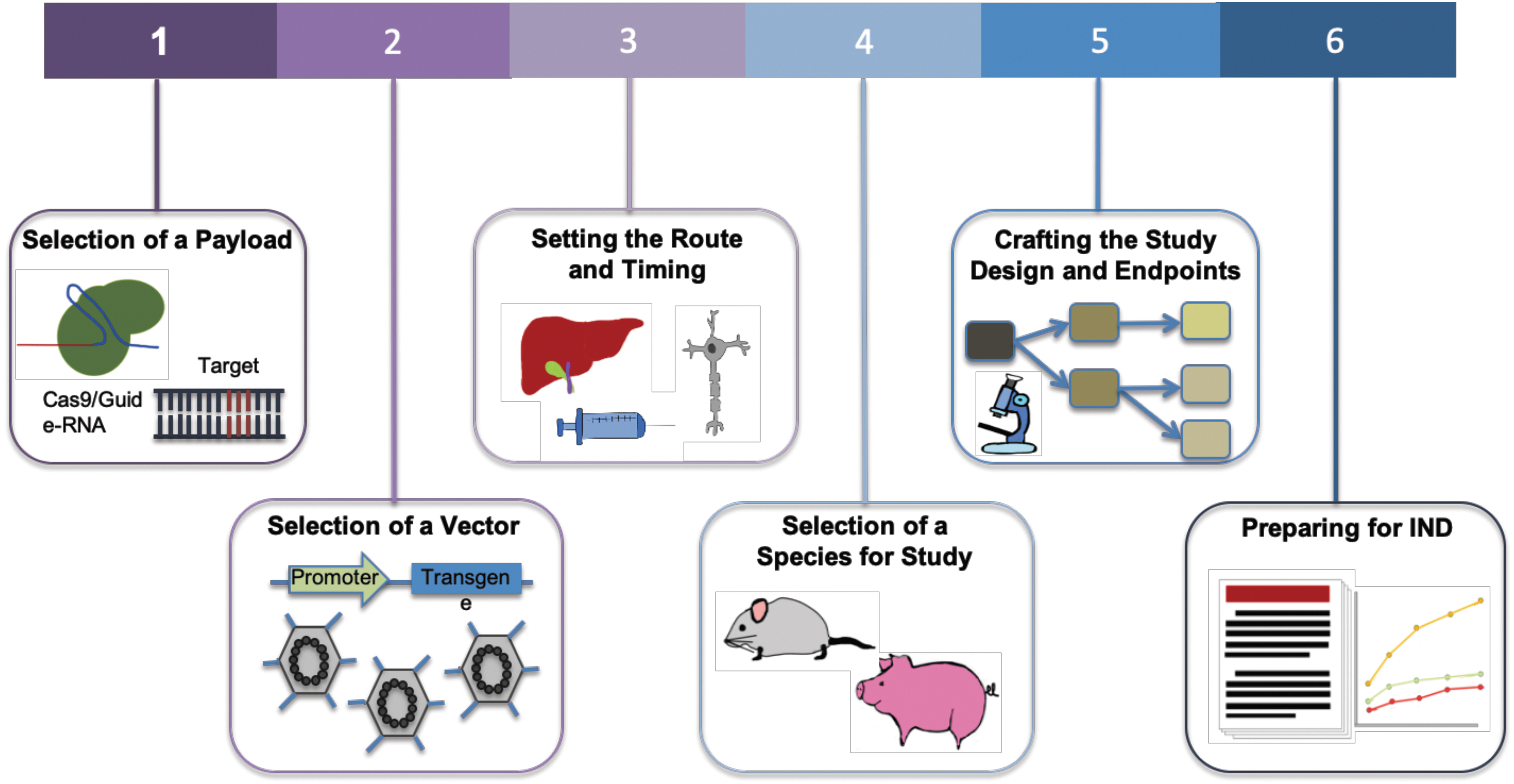
Steps toward enabling a gene therapy via CRISPR-Cas. For a disease target, the choice of Cas effector and corresponding gRNA is the first step. Next is to determine CRISPR-Cas delivery options via viral or nonviral methods. Associated with delivery system is also the route of administration. This typically depends on the organ or tissue of treatment and ideally should be limited to that location. Studies are then performed to assess the distribution, persistence, and clearance of the vector and the expression of the therapy in target tissues. Relevant clinical insight is also dependent on the selection of species for study, and a preclinical study design is crafted. Clinical endpoints are set based on safety and toxicology studies. Proof of safety and efficacy in preclinical studies will then lead to an IND application. gRNA, guide-RNA; IND, Investigational New Drug.
(1) Payload: The selection of payload is governed by the nature of the desired edit and considerations of enabling activity at the target locus while ensuring high specificity. The desired therapeutic correction determines the choice of nuclease, editor, or epigenetic modifier. Delivery constraints such as vehicle capacity and interaction with human biology and the immune system, availability of appropriate protospacer-adjacent motifs (PAM) sites, and the dynamic range of therapeutic effect also affect this choice.
(2) Delivery vehicle: Selection of a vehicle for in vivo delivery. Adeno-associated viruses (AAVs) are commonly used as they deliver the payload without insertion into the host genome, maintaining the packaged DNA in an episome. The AAVs work well in postmitotic cells and can be targeted via use of tissue-specific promoters or the selection of appropriate serotypes. 10 Efforts have been made to enhance the safety profile of AAVs by understanding how the viruses interact with and evade host immunity11–14 and engineering new capsids to mitigate the immune response. 15 Although AAV capacity limits the size of packaged constructs to about 5 kb, recent studies have uncovered Cas proteins that are considerably smaller than Streptococcus pyogenes Cas9, some of which show functional promise.16,17 Besides AAV, CRISPR-Cas can also be delivered via nonviral vehicles, with purified ribonucleoproteins (RNPs) and lipid nanoparticles (LNPs) showing promise for therapeutic translation.
(3) Delivery route and timing: Treatments delivered to the brain, spinal cord, and eye benefit from their immune-privileged status, and thus they are usually more tolerant to foreign antigens used in gene therapies. 18 For this reason and also the relative ease of delivery, some of the first clinical trials of CRISPR-Cas9 therapies have been initiated against hematopoietic cells where delivery is administered ex vivo or ocular disorders where delivery is via a subretinal AAV injection (NCT03872479). However, this immune privilege can also be breached when the blood–brain barrier (BBB) is compromised in certain pathological states.
The timing of the administration is also key, as studies have shown that the earlier the treatment is given in the course of disease progression, the higher the potential for efficacy. 19 Associated with this are studies to assess the distribution, persistence, and clearance of the delivery vector and expression of the therapy in target tissues. 20 A challenge to gene therapies can be the occurrence of off-target tissue effects, where expression occurs in unintended tissues or organs (in contrast to off-target genetic effects where edits occur at unintended genomic locations within the target cells). This can be minimized when therapies are administered to confined spaces, such as the eye or joints, 21 and through the incorporation of tissue-specific promoters to drive gene expression. 22
(4) Animal model: The fourth consideration is the selection of an appropriate animal model to obtain clinically relevant insights into the efficacy of the therapeutic. There are several challenges associated with testing gene therapies in animal models, including replication of human diseases, response to therapy, and differing immune reactions. However, as a result of advances in genome editing technologies, many animal models with specific alterations that are able to replicate clinical phenotypes have been generated. 23 In addition, the use of human immune system mouse models has improved the translatability of animal studies to the human setting. 24
(5) Study design: Multiple factors need to be considered when planning a preclinical study, including appropriate controls, number of animals per condition, testing across different dosages, and the times at which samples are taken. 25 When utilizing viral vectors for delivery, gene expression can begin after 1–2 weeks, and extend for months, during which samples are assessed. 26
Studies must also set endpoints and conduct measurements of clinically relevant phenotypes to assess the treatment's safety and efficacy. Toxicology studies establish potential local and systemic toxicities, safety and feasibility of the delivery system and procedure, immune response against the vector and delivered construct, and the potential for reproductive toxicity. Efficacy is examined via biodistribution studies, whereas clinical signs relevant to the disease are measured. 20
(6) Investigational New Drug preparation: An IND application can be prepared once preclinical experiments are analyzed, demonstrating safety and efficacy of the therapy. This application contains information about the animal pharmacology and toxicology, manufacturing and clinical components, and materials and protocols, 25 and it is submitted to regulatory agencies as a part of advancing into clinical trials.
Next, we discuss CRISPR-Cas-based gene therapeutics development in the context of this cascade. We highlight the progress made so far, as well as discuss pending challenges and approaches to overcoming them.
Challenges in Developing CRISPR-Cas Therapeutics
Payload
Among the most likely candidates for translating CRISPR-Cas components into functional therapeutics are the RNA-guided DNA nucleases, Cas9 and Cas12a; the RNA-guided RNA nucleases RCas9 and Cas13; and variants of these proteins. Different therapeutic modalities have been built with orthologs and variants from these Cas families, which together unlock a variety of clinical applications. The native nuclease function of Cas9 or Cas12a can be directly applied to genome editing. The resultant double-stranded DNA break is repaired by native cellular machinery, typically through the nonhomologous end-joining pathway27–30 or homologous recombination with an endogenous sequence or an exogenously provided sequence. This can be applied to engineer gene knockouts, edit a regulatory region or transcription factor-binding site, create large deletions, or enable gene repair or replacement. Further therapeutic approaches toward genome editing with Cas9 have been demonstrated with the advent of base editing and prime editing, with the former more efficiently used for single-base transitions and the latter showing considerable potential for a wider range of localized edits, although with lower efficiencies.
Targeting diseases at RNA level is appealing as it prevents any permanent off-target effects, making it less risky for clinical applications. RNA-targeting RCas9 and Cas13 have been used for RNA knockdown with a similar or comparable efficiency to RNA interference (RNAi).31,32 Common RNA-editing strategies rely on naturally occurring human adenosine deaminase acting on RNA (ADAR) enzymes. Modification of ADAR2 and fusing it to a catalytically inactive Cas13, known as RNA editing for Programmable A to I Replacement (REPAIR), recognizes and corrects disease mutations without any PAM sequence constraints. 33 There is increasing interest in developing Cas-mediated transcript editing into RNA therapeutics with applications in treating viral infection or alleviating symptoms in neurological disease.34–36
Together, these CRISPR-Cas modalities enable a broad spectrum of strategies toward disease treatment (Fig. 2).
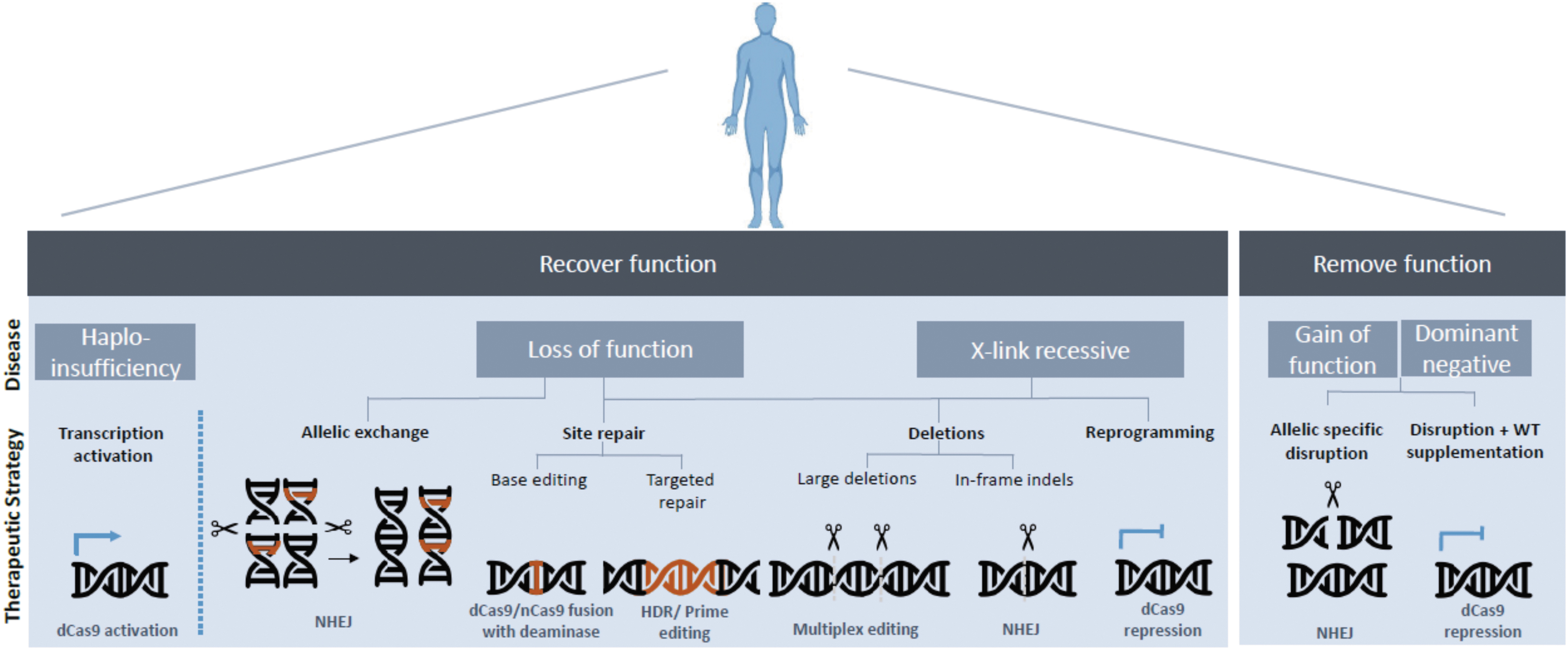
CRISPR-Cas therapeutic strategies toward disease-causing mutations.
Delivery
Delivery of the CRISPR-Cas therapeutic cargo represents one of the key challenges in developing a clinically viable CRISPR-Cas therapy.
CRISPR-Cas systems can be delivered in three cargo formats: via DNA modalities encoding both the Cas protein and gRNA (such as plasmids, DNA viruses, nanoparticles); RNA modalities encoding the Cas and the gRNA (modified RNA, RNA viruses, in vitro transcribed RNA, nanoparticles); or direct delivery of the purified Cas protein complexed with the gRNA as an RNP or nanoparticle. Each of these cargo formats has been used ex vivo (in cells outside the body) and in vivo (locally within specific tissues or systemically through the body) (Fig. 3). Many of these delivery methods are built on the established framework of traditional gene therapy and, hence, share many of the technological underpinnings and desired properties.
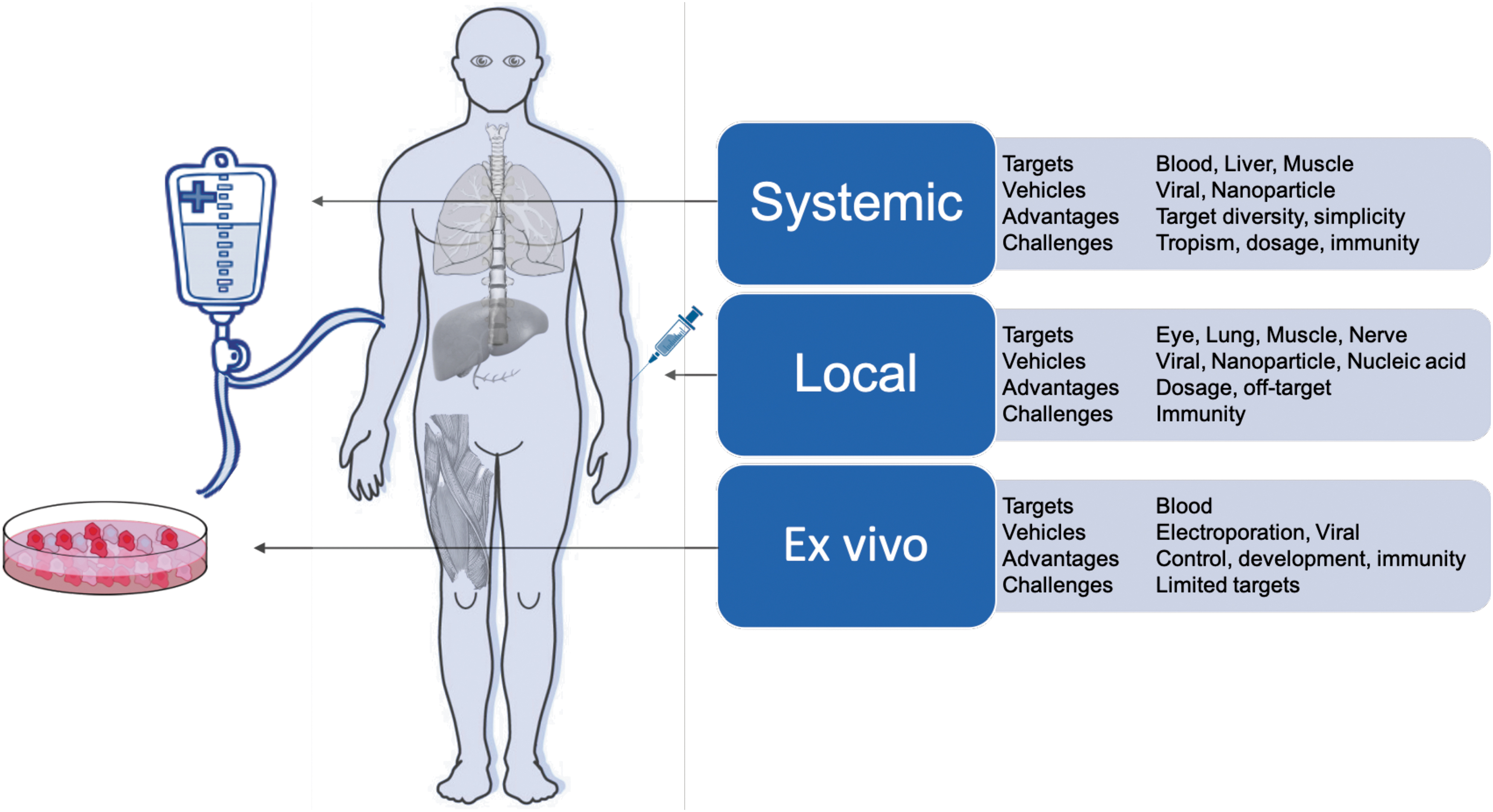
Delivery modalities for CRISPR-Cas therapeutics.
The ideal delivery system will likely encompass the following attributes: (1) nonintegrating: limiting unintended integration events in the genome; (2) specific: achieving targeted tissue delivery; (3) low immunogenicity: enabling low toxicity associated with administration and sustained therapeutic effect; (4) option to re-dose: allowing repeated regimens; (5) biodistribution: compatible with target tissue type and tissue accessibility; and (6) scalable: compatible with large-scale manufacturing for both common and orphan diseases. We describe later how these different delivery modalities have been used for CRISPR-Cas and how they might exhibit some of these ideal attributes.
Ex vivo delivery modalities
Ex vivo gene editing allows cell engineering and quality-control regimes that would not be feasible within the human body. It is most compatible with hematological disorders, as blood is much easier to safely remove, treat, and re-infuse than other tissues. A modality successfully used for ex vivo gene editing is Cas proteins complexed with sgRNAs in vitro as RNPs. Unlike DNA- or RNA-encoded CRISPR, RNPs do not rely on cell-driven expression and hence have the quickest onset of activity on administration. In addition, RNPs are not further expressed and degrade over time, offering a transient window of exposure that limits potential off-target effects. 37 The most clinically advanced CRISPR-Cas editing strategy relies on ex vivo RNP-mediated editing followed by re-administration of the cells back to the donor.
Advances in the isolation and propagation of hematopoietic stem cells (HSCs) and immune cells have promoted cellular gene therapy as a viable option for the treatment of some monogenic diseases and malignancies. Early successes were complicated by uncontrolled insertional mutagenesis of the vector, resulting in transcriptional activation of nearby proto-oncogenes, chromosomal translocations, and eventual leukemogenesis. 38 Unlike traditional gene therapy requiring sustained transgene expression, CRISPR-Cas systems can achieve therapeutic outcomes by transient expression, allowing for less integrative risk and potentially safer methods of delivery. Current ex vivo CRISPR-Cas mediated clinical trials utilize direct delivery of Cas9 RNP via electroporation (NCT03399448, NCT04035434, and NCT03655678).
Curative approaches for β-hemoglobinopathies include allogenic hematopoietic stem cell transplant (HSCT) by using HLA-matched stem cells derived from a donor or autologous transplantation of the patient's own HSCs after receiving the gene therapy ex vivo. Allogenic transplantation is challenging due to dependence on finding an immune-matched donor and mitigating the effects of graft-versus-host response. Autologous transplantation with gene-edited cells avoids much of this immune barrier. Although many indications require varying levels of conditioning treatment to reduce the population of native HSCs to create space for donor cells, autologous ex vivo gene therapy may also allow for a reduced intensity of this process for patients. 39 The current approach toward HSC gene therapy uses electroporation of CRISPR-Cas mRNA or RNPs to edit HSC genomes. 40 If homology-directed repair is required, the template is simultaneously electroporated or transduced by viral vectors. 41 Following this approach of ex vivo editing and re-transplantation into the body, the first two patients with β-thalassemia and sickle cell disease were treated in 2019 by using CRISPR-Cas9-edited HSCs and autologous transplantation.
The RNP-based approach has also been successful in progressing toward clinical trials. For instance, Cas9 RNPs were electroporated into HSCs to disrupt BCL11A, a silencer of fetal hemoglobin, to generate CTX100 cells. Disruption of BCL11A de-represses fetal hemoglobin expression and rescues sickle-cell defects.42,43 Another therapeutic candidate, CTX110, utilized the multiplexing capability of CRISPR-Cas systems to edit CAR-T cells at multiple loci. This approach results in replacement of the native T cell receptor (TCR) with an anti-CD19 CAR as well as knockdown of MHC I expression via targeting β2M (NCT04035434). Positioned as “off-the-shelf” therapy, using allogenic donor T cells could reduce the cost of single batch production. Another ambitious study used Cas9 RNPs to disrupt PDCD1, TRAC, and TRBC, and it introduced transgenic NY-ESO-1-specific TCR to generate engineered T cells for a clinical trial last year. 5 The controlled environment of cell engineering and quality control (QC) relative to in vivo administration is the reason that ex vivo pipelines have been among the first to progress to the clinic, and it will inform the uptake of genome editing for in vivo use.
In vivo delivery modalities
Adeno-associated viruses
Viral vectors are the most common in vivo delivery vehicles for CRISPR-Cas. Viruses have substantial differences in their safety profiles, with AAVs having one of the more favorable profiles. The recently approved gene therapies Luxturna 3 and Zolgensma 4 deliver their therapeutic transgenes via AAVs. Soon after, the first in vivo CRISPR-Cas therapy entered a clinical trial, in which AAV5-CRISPR-Cas9 was delivered into the eye for safety and efficacy evaluation (NCT03872479). These clinical and commercial successes make AAVs an attractive choice for upcoming therapeutic development pipelines.
AAVs are replication-defective viruses that naturally infect humans and nonhuman primates (NHPs). After infection, the DNA carried by AAVs achieves long-term episomal expression. AAVs are endemically found and have several variants that exhibit different tropism for various tissues, such that a serotype can be selected to best target the tissue and disease indication of interest. 44 Significant work has been devoted to AAV capsid protein engineering. For example, rational capsid engineering has been used to increase neuronal transduction by mutating surface-exposed tyrosine residues and phosphorylating threonine residues on the AAV2 capsid. 45 Altering vector capsids can also increase tropism toward tissues that are challenging to target, such as microglia. 46 Chimeras with new functions can be derived from peptide-domain exchanges among different serotypes, such as AAV2.5, which has improved muscle transduction. Altering the immune profile away from both parental AAV1 and AAV2 serotypes offers a potential strategy for immune bypass and therapeutic re-dosing. 47 More complex chimeric capsid libraries can be generated by DNA shuffling of capsid sequences of multiple AAV serotypes to generate greater variation and accelerate the evolutionary process. 48 Novel capsids have also been generated by using polymerase chain reaction (PCR)-based mutagenesis and subsequent selection for specific attributes. 49 For instance, directed evolution has been an efficient approach to derive tissue tropisms in induced pluripotent stem cells (iPSCs) 50 and human ciliated airway epithelium. 51
Viral protein engineering has also been used to improve the safety of gene therapy vectors. Even though infection with AAVs is only mildly immunogenic and not associated with disease, the remaining immunogenicity still represents a significant safety and efficacy challenge. The effective use of AAV vectors can be compromised by a pre-existing immune response from natural exposure to AAVs in early life or from prior AAV-mediated therapy.52,53 Adaptive immune response to AAVs leads to production of neutralizing antibodies (NAbs) that block transduction and clear the viruses, whereas adaptive cellular response by CD8+ cytotoxic T lymphocytes (CTL) leads to the eradication of transduced cells and can attenuate treatment efficacy and impose safety risks. One strategy used to overcome these issues involves engineering the AAV capsid epitopes to avoid antibody and T cell recognition. Immune-reactive epitopes have been mapped for some serotypes, and capsid mutagenesis can reduce binding and neutralization effects.54,55 However, it is not always straightforward to engineer epitopes while maintaining desired functionality. Often, such modifications result in loss of function, as exemplified by a study where an AAV9-specific neutralizing epitope was mapped to capsid residues conferring liver tropism; unfortunately, mutations did not provide a successful tradeoff in evasion from polyclonal antibodies. 56
In addition to induced or pre-existing adaptive immune responses, AAVs trigger a natural innate immune response through recognition of pathogen-associated molecular patterns inherent to AAV biology. 57 Multiple studies have elucidated some of the mechanisms of innate immune activation by AAVs, including binding by TLR9 of unmethylated CpG dinucleotides in the AAV genome, leading to activation of the MyD88/NF-κB pathway,58–60 and recognition by MDA5 of long dsRNAs created by bidirectional transcription from the AAV episome. 61 These pro-inflammatory signals result in substantial transcriptional changes, inducing an antiviral state within the target cell, and expression of immune-stimulating cytokines that allow for a robust adaptive immune response. 62
In the clinic, patients' cytokine levels are routinely monitored, and sometimes they are modulated with corticosteroids. 63 In contrast to other viral vectors, AAVs have not demonstrated a clinically dangerous level of innate immune activation in humans, although certain cell types may be susceptible to damage, thus reducing clinical efficacy. 64 Nevertheless, minimizing innate immune induction remains an important goal in AAV vector biology to maximize patient safety and limit adaptive immune induction.
More recently, AAV engineering has expanded beyond protein engineering to include chemical and biophysical methods. Chemical modification introduces synthetic motifs to the AAV surface. An early chemical approach masked exposed arginine residues by a naturally occurring glycation reaction. 65 Recently, a site-specific approach was developed by using unnatural amino acids on AAV capsids to attach synthetic ligands through biocompatible small-molecule chemical reactions termed “click chemistry.” This strategy was used to tether ligands on the AAV capsid to achieve tissue-specific targeting and protection against antibodies.66,67 Another approach involves encasing the AAV in a membrane exosome, allowing for greater customization of the external layer of the drug. Exosome-enveloped AAV vectors achieved enhanced transduction and protection against pre-existing neutralization antibodies.68–71 A recent study demonstrated the stabilizing effects of tetraspanin CD9 in increasing exosome production. 72 Although it is an attractive solution, manufacturing methods for scalable production of exosomes will be challenging.
Nanoparticles
Nonviral delivery systems are also very promising. Various nanomaterials such as polymers, lipids, proteins, and metals have been explored for in vivo delivery. Following extensive research on LNPs, there are currently more than 10 FDA-approved uses of LNPs for drug delivery. 73 Onpattro is a recently approved multi-dose gene therapy utilizing LNP to deliver small interfering RNA for the treatment of polyneuropathies.
LNPs typically comprise cationic lipids that facilitate endocytosis and are released into the cytoplasm and PEGylated lipids that facilitate stabilization and prevent nonspecific interaction. 74 PEGylation also reduces immunogenicity and increases circulation time. 75 Recently, an LNP-based delivery system incorporating helper lipid and PEG-DMG to encapsulate Cas9 mRNA and modified sgRNA was able to produce durable editing of ∼70% in the liver, benefiting from a multiple dosing regimen. 76 Other studies targeting the liver by LNPs showed robust knockdown 77 and an HDR-editing rate of 6%. 78 Beyond the liver, however, applications might be more limited: Lipid particles are usually taken up and metabolized in the liver during systemic circulation, 79 thereby constraining the targetable tissue types for LNPs.
Gold nanoparticles can be used to bind Cas9 RNPs and conjugate with 5′-thio ssDNA for further hybridization with DNA repair templates. 80 The nanoparticle is then coated with negatively charged silica for cationic polymer PAsp(DET) encapsulation, after which cytoplasmic glutathione releases the contents from the gold nanoparticle. 81 Intramuscular injection of CRISPR Gold Cas9 RNP with template DNA was able to correct 5.4% of dystrophin gene mutations. Intracranial injection of CRISPR Gold Cas9 RNP resulted in localized gene editing of 14.5% of the GRM5 gene and restored 40–50% of the protein's production in the brain and other tissues, rescuing the effects of ASD in a mouse model. 82 The gold nanoparticles are well tolerated in the neurons; however, the long-term accumulation and elimination kinetics of these nanoparticles remain to be evaluated.
Ribonucleoproteins
Beyond use as delivery vehicles, established protocols for large-scale production of proteins make direct delivery of the Cas9 RNP complex an attractive clinical proposition. Many polymers are being evaluated for coupling with the Cas9 RNP complex, which has a heterogeneous charge distribution. A mix of cationic and anionic monomers with imidazole can fully encapsulate the RNPs while facilitating endosomal escape. The glutathione cleavable link N,N′-bis(acryloyl)cystamine around the RNP enables cytosolic release in the presence of glutathione, but to boost the efficacy of delivery and therefore editing, a further attachment of tissue-specific ligands to one end of the PEG is necessary. 83 Although poly(aspartic) acid (PAsp) based polyplexes are biocompatible with limited toxicity, modified PAsp(DET) bearing 1,2-diaminoethane side chains show higher transfection efficiency. 84 Recently, PAsp(DET) assembled nanoparticles were able to effectively deliver Cas9 RNP complexes into muscle and brain tissues.80,82 A screening effort also identified poly(aspartic) acid polymer (PAsp) analogs to efficiently deliver Cas12 RNP in mice muscle fibers. 85
Administration
Manufacturing and scale-up challenges
In general, manufacturing of pharmaceutical agents must conform to current Good Manufacturing Practice (cGMP). This often refers to a set of regulatory laws in the U.S. FDA Code of Federal Regulations (CFR) title 21, although other countries and organizations, particularly the WHO and EU, have defined their own similar standards for cGMP. Although the application of GMP may vary depending on a wide range of pharmaceutical agents and production methods, generally it requires the use of dedicated clean production facilities, controlled starting materials and cell lines, and multiple product purification steps with accurate testing at each step. cGMP regulation is often focused on process controls and extensive documentation such that a wide variety of issues, should they arise, can be easily detected and corrected. The components of a cGMP production system will vary depending on the agent being produced and must be evaluated on a case-by-case basis.
Nucleic acid manufacturing
The foundation for generating CRISPR therapeutics is nucleic acid, either as synthetic gRNA, a DNA template for gRNA transcription, plasmid encoding the Cas effector protein, or plasmid encoding delivery vehicle components, for example, viral vectors. The manufacturing specifications required for these nucleic acids may differ depending on the application, but they will generally need to conform at minimum to high quality (HQ) grade manufacturing for vector plasmids and template DNA not used in the final therapeutic, and ideally to full cGMP grade for direct applications.
HQ grade DNA can be produced in dedicated facilities by using strains drawn from a research cell bank, utilizing two to three chromatography steps to eliminate linear, open, and chromosomal DNA and remove LPS and other cellular contaminants. The main differences when upgrading to cGMP production are the necessity of a GMP cell bank starter, a dedicated GMP facility, a validated quality assurance system with in-process control, and full documentation procedures (For a more thorough discussion of these procedures and QC validation requirements see Ref. 86 ).
Despite these stringent requirements, the production of cGMP-compliant, therapeutic-grade nucleic acid is much easier than recombinant protein. Fewer purification steps are required, as the reaction mixtures are much less complex than those involved in bacterial or mammalian cell culture. In addition, similar to protein therapeutics, the production of DNA- and RNA-based therapies has advanced rapidly in the past two decades due to excitement surrounding the potential for nucleic acid vaccines and RNAi drugs. Utilizing many of these same facilities and procedures, several companies offer cGMP preparations of sgRNA for clinical and preclinical CRISPR research, as well as HQ and cGMP viral vector preparations. The cost for these materials is manageable, except for particular cases requiring truly massive quantities of DNA, such as low-yield AAV vectors needed for systemic delivery. Nucleic acid production with cGMP guidelines are a feasible and preferred option for more typical RNP and high-yield AAV serotype delivery modalities.
Manufacturing of nonviral RNPs
The current nonviral formulations for CRISPR-Cas range from simple to difficult for manufacturing, with each carrying its own particular set of challenges for creating therapeutic-quality products. The most clinically advanced nonviral delivery modality is also one of the simplest to produce, with electroporated RNPs consisting of only two purified components, namely a gRNA and a Cas protein. The in vitro transcription or de novo synthesis of gRNAs is well established. Cas proteins such as Cas9, Cas12a, or engineered variants would be recombinantly produced via scalable fermentation in pipelines similar to thousands of other protein therapeutics. After production in fermentation vessels, the resultant product is run through multiple chromatography steps to concentrate the active product and remove impurities.
Much of the initial recombinant protein production for pharmaceuticals was dominated by classic producers such as Escherichia coli and Saccharomyces cerevisiae. 87 Since then, most biopharmaceutical production has been deployed by using mammalian cell lines, especially Chinese Hamster Ovary cells or human cells, due to the superior protein-folding intracellular environment and post-translational modifications. 88 Still, microbial production retains an important niche owing to its advantages in rapid growth, low nutritional requirements, and ease of genetic manipulation. 89 Indeed, Cas effector protein production seems to fit nicely into this niche based on their bacterial origin and independence of complex folding cofactors or post-translational modifications. Process improvements in bacterial production, such as the deployment of an endotoxin-free E. coli strain, 90 metabolic engineering efforts, 91 and adaptive laboratory evolution, 92 will yield additional improvements that are directly applicable to CRISPR-based protein therapeutics. Further, new exploration of alternative manufacturing methods, such as co-production of Cas9 protein and sgRNA in E. coli, may open up new avenues of inexpensive drug production. 93
Nevertheless, current manufacturing methodologies are poised to implement recombinant Cas proteins at scale. Commercial recombinant Cas9 preparations, such as Aldevron's SpyFi™ Cas9 nuclease, are available. Importantly, associated procedures should be equally amenable to production of other Cas9 effectors, including dCas9, Cas12a, and variants thereof. Pharmaceutical companies have been highly incentivized to optimize scaled protein production environments, 94 including moving to continuous flow rather than batch setups, 95 due to lucrative demand for protein therapies, especially monoclonal antibodies. Process advancements brought about by this economic landscape will translate well to CRISPR-based protein drugs.
The RNA component of the CRISPR-Cas RNP can comprise a synthetic crRNA and tracrRNA pair as in the native bacterial system, 96 a synthetic combined sgRNA, 97 or an in vitro transcribed sgRNA. 98 Single gRNA constructs are preferable due to simplicity and elimination of the duplex formation step. Both synthetic and in vitro transcribed gRNAs have associated benefits and drawbacks. Chemically synthesized gRNAs are amenable to modification of one or more of the nucleotide bases, which provides advantages such as increased stability 99 and reduced immunogenicity. 100 New modifications to further optimize these parameters are currently being explored.101,102 The potential advantages of in vitro transcribed gRNA production include simple and economical scale-up to large volumes, as the generation of templates can be done with PCR. Both in vitro transcribed and chemically synthesized gRNAs are being evaluated in ongoing clinical trials.5,103
AAV manufacturing
AAVs are the prime viral delivery vehicles being explored for in vivo CRISPR therapies given several beneficial properties, including low immunogenicity, nonintegration, different serotypes available for different tissue tropism, and a proven clinical record. Nonetheless, despite years of intense research and development, manufacturing challenges still exist.
Typically, AAVs are produced by using a triple transfection method in which plasmids encoding (1) the recombinant genome to be delivered, (2) the viral packaging genes, and (3) adenoviral helper genes are transfected into cells, most commonly human embryonic kidney (HEK)293 lines by using chemical agents such as polyethylenimine. Many AAV products for clinical trials have been produced by using these methods (NCT02122952, NCT25322757, NCT21031578, NCT27453480). Initially, this was done by using adherent HEK293 cells cultured in 2D on stacks of flasks. These formats can suit typical doses for early trial phases that range from 1011 to 1013 vector genomes (vg) per patient. However, achieving titers high enough for later phases or larger doses would mean hundreds of flask stacks and many labor months. 104 This obstacle is most pressing for systemic disease targets such as Duchenne muscular dystrophy (DMD), which require larger doses to achieve good efficacy. Current recommendations for clinical doses are as high as 1015, even 1016 vg per patient. The product volumes required for clinical trials, especially critical phase III trials with large patient cohorts, are simply impractical by using traditional cell culture plasticware.
As a result, both academic and industrial facilities have also adopted more scalable platforms to produce AAVs, particularly in fixed-bed bioreactors with adherent HEK293 cells 105 and bioreactors with suspension HEK293 cells. 106 Both have provided scalability without a complete process overhaul. Still, yields using these methods have largely been unable to exceed 1014 vg per liter, necessitating thousands of liters for larger clinical trials. In addition, scaling up requires nontrivial quantities of GMP-grade plasmids, which face scalability problems of their own. 107 One approach to the transfection scalability issue is to deliver the necessary genetic material by using viral carriers, typically a herpes virus or baculovirus, rather than by chemical transfection. 108 This approach has led to a moderate increase (twofold to fivefold) in yield, 109 and interestingly, an increase as well in AAV product infectivity in the case of herpes virus production, potentially due to superior genome loading into AAV capsids in the presence of helper genes from the herpes virus. 110
This yield challenge has also prompted the use of baculovirus-infected insect Sf9 cells for AAV production, which provides a much higher yield than HEK293 and has been used in multiple clinical trials and production campaigns.111–113 Nonetheless, a recent revelation that Sf9-produced AAVs differ qualitatively from HEK293-produced AAVs creates a potential roadblock that requires further investigation. 114
Additional developments, including continuous flow-based production methods and optimization of producer cell lines, may further increase AAV yields. Still, all AAV production methods require major downstream processing, including multiple filtration steps, size exclusion chromatography, ion exchange chromatography, and ultracentrifugation, to achieve the required purity and enrich for properly packaged, infectious vectors.112,115 Even applying numerous recent technical advancements, scalability of AAV vector production remains a huge challenge, especially for relatively common diseases requiring large effective doses such as DMD.
Pre-IND and preclinical development of CRISPR-Cas therapeutics
Successful gene therapies require preclinical validation in animal models. Different preclinical models have been developed to collect data in three main areas: proof of concept for efficacy, determining dose ranges for the chosen route of administration, and evaluating toxicity and safety profiles. For AAV-CRISPR-Cas, four main organs have been targeted most frequently and successfully. This initial success builds on more some three decades of understanding in gene therapy and will itself guide future CRISPR therapeutic development.
Muscle
There is significant morbidity and mortality in hereditary disorders of the muscle, and gene therapy offers promising outcomes for these unmet needs. Many studies have demonstrated the effectiveness of using skeletal muscles as protein factories to express transgenes, contributing to the first gene therapy product (Glybera) being officially approved. 116
DMD is one of the most common hereditary muscular diseases in children and is caused by mutations in dystrophin, the largest gene in the human genome. Progress in gene-editing technologies has raised hopes for successful restoration of the dystrophin gene. Conventional gene therapy has been challenging due to the AAV packaging limit; hence, efforts have focused on truncated versions of dystrophin for gene replacement, such as microdystrophin. The therapeutic strategy of AAV-mediated gene therapy in muscles appears safe in preclinical and clinical studies, but so far the therapeutic effects have been limited in DMD individuals. 47 Patients with large frameshift deletions also risk T cell-directed destruction against the new epitopes.117,118 The lack of long-term effects could also be due to constant muscle degeneration and regeneration, diluting the expression of the nonintegrated transgene with time. Another strategy is the use of CRISPR-Cas to permanently restore endogenous dystrophin expression. Diverse CRISPR-Cas gene-editing strategies have been employed to treat muscular dystrophy mouse models, including exon skipping, exon deletion, adenine base editors, and HDR (Table 1).119–122
AAV, adeno-associated virus; ALS, amyotrophic lateral sclerosis; APP, amyloid precursor protein; Bace1, beta-secretase 1; DMD, Duchenne muscular dystrophy; FIX, factor IX; IDUA, α-
As DMD affects muscle groups throughout the body, the challenge is to develop a single injection that is sufficient for whole-body therapy. Treating DMD systemically via intravascular injection allows targeting of all muscle groups and has been shown to be less immunogenic. 123 Durable levels of dystrophin were detected in cardiac and skeletal muscles in systemically treated neonatal mdx mice with AAV-SaCas9. 124 Moreover, systemic delivery of AAV9-SpCas9 and AAV9-sgRNA corrected frame-shifted mutant DMD, restoring up to 90% dystrophin expression in a canine model of DMD. 125 However, intravascular administration will require high doses of AAV to reach therapeutic levels of gene editing in clinics. The Sarepta Therapeutics clinical trial NCT03375164 efficacy dose is 2 × 1014 vg/kg, where a single treatment of a patient will require 1015 vg of AAV. One approach to reduce AAV dose is the use of self-complementary AAV (scAAV), which in one study has shown up to 20-fold dose reduction. 126 However, the large payload requirements with CRISPR-Cas raise limitations on the potential use of this strategy.
Eye
Gene therapy in the eye is attractive due to the anatomical compartmentalization that enables targeting of specific cells with minimal exposure to other organs and the immune system. Specific cell types in the eyes such as retinal pigment epithelium, ganglion, and photoreceptors can be effectively transduced by AAVs, and surgical processes afford access to the various structures in the eye. 127 Non-invasive methods have been established for evaluating therapeutic effects, whereas the contralateral eye can be used as a convenient control. Lastly, numerous genetic animal models have been generated for diseases in the retina, paving the way for gene therapy development. 128
Leber congenital amaurosis (LCA) is a childhood-onset blindness caused by mutations in at least 1 of 18 different genes. Type II is the most studied form, with mutations in the RPE65 gene. Gene replacement of RPE65 in a blind canine model demonstrated improved vision through simple behavioral tests. More importantly, the therapy demonstrated cortex recovery and response to visual stimulation despite prolonged visual deprivation in the RPE65-mutant dogs.129,130 The results suggested safety of the AAV2 subretinal injection and some level of vision restoration when RPE65 was first introduced in LCA patients.131–133 Re-administration of the therapy in the second eye was also successful in large animal studies despite elevated serum NAb toward AAV2134,135 and proved predictive of the results in human clinical trials. 136 AAV2 transfer of RPE65 showed therapeutic efficacy in phase III randomized trials and is marketed as Luxturna.
After the landmark success of Luxturna, the first in vivo CRISPR-Cas gene editing human trial program was developed by Editas Medicine to treat LCA type 10. In this case, subretinal injection of AAV5 delivering SaCas9 is driven by a human rhodopsin kinase (hGRK1) promoter to repair the splicing defect of CEP290. hGRK1 promoter is able to drive robust transgene expression in both cones and rods in NHPs and mice.137–139
A wave of new treatments is being developed for inherited retinopathies. In particular, the CRISPR-Cas editing approach is attractive for retinitis pigmentosa (RP) as 25–30% of the cases are due to autosomal dominant mutations. 140 CRISPR-Cas mediates allele-specific editing by recognizing the unique PAM derived from point mutations in dominant disorders such as RP and Meesman epithelial corneal dystrophy.141,142 However, many disease mutations such as RHO P23H do not create a novel PAM. One study improved allele discrimination in heterozygous Rho P23H mice by placing a single-base mutation in the spacer region of the sgRNA coupled with the SpCas9 VRQR variant. 143 Interestingly, another study reported that increasing wild-type to mutant Rho expression was able to slow retinal degeneration. 144 This suggests that therapeutic benefits can be attained despite incomplete mutant disruption and even with some indiscriminate wild-type downregulation.
Chronic eye diseases such as age-related macular degeneration (AMD), the leading cause of blindness, are another prime target for gene therapy. Current treatment requires regular anti-VEGF intravitreal (IVT) injections but with only modest improvements in patients with advanced neovascular/wet age-related macular degeneration (Wet AMD). Previous clinical trials using AAV2-sFLT01, expressing soluble VEGF receptor in Wet AMD patients, showed variable results, 145 so other strategies have been developed to reduce angiogenesis. Clinical trial NCT03748784 is using an IVT injection of AAV7 m8-expressing aflibercept (anti-VEGF protein). In addition, a recent CRISPR-Cas approach targeting VEGF-A using AAV8-SpCas9 suppressed 31% of laser-induced choroidal neovascularization in mice. 146 Overall, the pathogenesis of AMD is complex when compared with monogenic disorders and a multimodal approach enabled by CRISPR technologies might be beneficial.
Successful gene therapy treatment is also dependent on effective administration at the target tissue and the resultant immune response. Subretinal injections are efficient for targeting photoreceptors and RPE, whereas IVT injections target cells in the inner retina such as ganglion cells. 147 Although IVT administration of AAV provides a broader tissue application in the retina, there is an increased likelihood of generating NAbs against the AAV capsid and transgene as shown in NHPs. 148 In addition, T cell responses against AAV2 capsid were also detected during IVT injection in an NHP toxicology study. 149
Altogether, studies show that IVT administration increases AAV exposure to the immune system. 150 Another consideration is the administration complexity and ensuing complications. The IVT injection is a routine procedure for drug delivery whereas the subretinal injection is more invasive, increasing risk of complications. 151 Therefore, multiple studies have tried targeting RPE by using different AAV serotypes via IVT administration. Much research has been done to improve AAV transduction of photoreceptors by AAV capsid engineering or to reduce physical barriers by surgical methods such as vitrectomy or by peeling of the internal limiting membrane.152–154
Liver
The liver is an essential organ that metabolizes and synthesizes intracellular and secreted proteins. Defects in lipid and protein metabolism or detoxification due to liver disease are often destructive to multiple organs, causing complex disorders. Liver-directed gene therapy is an attractive option in the treatment of metabolic diseases, demonstrating safety in acute intermittent porphyria clinical studies as well as monogenic diseases such as hemophilia B.155,156 Hepatocyte-directed gene transfer is further enabled by easy access of the AAV vector through liver sinusoids, as it is small enough to pass through the fenestrae. 157
In the treatment of hemophilia and other monogenic diseases, AAV vectors are preferred for liver-directed gene therapy as nonviral and retroviral delivery methods only result in transient expression.158,159 The earliest in vivo AAV vector used was AAV2, which has broad tissue tropism, including hepatocytes. Subsequently, AAV8 was shown to be 10- to 100-fold more efficient in transducing hepatocytes.160–162 Further, humans have a lower prevalence of NAb against AAV5 and AAV8 compared with AAV2,163,164 resulting in a shift favoring AAV8 and AAV5 serotypes for liver-directed gene therapies. Liver gene therapy clinical trials have been tested for a small number of diseases, with hemophilia B studies taking center stage. 14
Ultimately, hemophilia patients need to maintain at least 1% plasma circulation for effective hemostasis in their lifetime. AAV-mediated transfer of factor IX (FIX) or factor VIII (FIIIV) in liver, especially in children, will result in dilution of the transgene as the hepatocytes divide. This problem can be mitigated by site-directed genomic integration. A gene-replacement strategy using CRISPR-Cas to integrate the transgene into the albumin locus downstream of the endogenous albumin promoter produced sustained FIX and FIIIV expression in hemophilia mouse models.165,166 AAV8-Cas9 mediated integration of human FIX-padua exon 2–8 in exon 2 of mFIX and a codon-optimized FIIIV into intron 13 of albumin, and it developed persistent FIX or FIIIV levels for 7–8 months with no complications.165,166
A similar strategy proved effective in the treatment of ornithine transcarbamylase (OTC) deficiency, an X-linked urea cycle disorder where a codon-optimized human OTC was inserted into the intron 4 of mouse OTC locus by using AAV8-SaCas9. 167 Conversely, Cas9-mediated HDR to correct the mutation had resulted in large deletions and affected endogenous OTC expression in the spf/ash heterozygous OTC mice. 168 Cas9 integration of OTC produced both rapid and prolonged gene expression when compared with conventional gene replacement therapy. 167
Preclinical studies have shown efficacy in liver-directed gene transfer for lysosomal storage diseases (LSDs) such as Gaucher disease, Fabry disease, and glycogen storage disease type Ib and II.169–172 LSDs are mostly monogenic and include more than 40 different metabolic diseases. The spectrum of disease severity also spans from disorders in the central nervous system (CNS) to systemic multi-organ pathology, as the enzymes are ubiquitously expressed. 173 Current treatments such as enzyme replacement therapy (ERT) and HSCT have limited efficacy, especially for those with CNS disease.174,175 Prolonged ERT is associated with poorer quality of life, and intravenous ERT has been insufficient to remedy neurological manifestations, 176 whereas HSCT is associated with transplant-related morbidity and mortality rates of 10%.177–179
Interestingly, recent studies in LSDs were able to bring about some neurological benefit by using liver-directed gene editing. The effects in CNS were likely due to enzymes crossing the BBB rather than gene editing in the brain, as editing was driven by a liver-specific promoter. This was demonstrated in AAV-SaCas9-mediated transgene integration into the albumin safe harbor locus in Sandhoff mice.
180
Sangamo Therapeutics, in a promising preclinical study, used an AAV2/8 zinc finger nuclease editing strategy to insert α-
Central nervous system
Neurological disorders are difficult to treat due to the complexity of the CNS. Although conventional therapy has limited access to the brain, specific AAV serotypes are able to cross the BBB due to their unique tissue tropism. 183 A noninvasive mode of delivery by intravenous injection of AAV9 can transduce the spinal cord, including motor neurons and astrocytes in mice, large animals, and NHP.184,185 Intra-cerebrospinal fluid (CSF) delivery is also able to provide widespread distribution in the CNS and reduce transduction of periphery organs.186,187 The third approach is to directly deliver to target regions, limiting therapy to the site of administration; hence, broader delivery will require multiple injections and surgery. 188
Zolgensma—the approved CNS targeting gene therapy for SMA—delivers scAAV9-SMN1 systemically to target motor neurons. 4 However, the high vector dose has contributed to an exorbitant price for the therapy. Alternatively, the dose can be reduced with intra-CSF administration by intracerebroventricular and intrathecal injections. Gene transfer in a severe SMA mouse model was able to reduce the dose by 10 times with intra-CSF administration to improve survival. 189 Safe and robust widespread transduction in the motor neurons of the spinal cord was also demonstrated by an intrathecal injection of AAV9 in pigs. 190 As a result, intrathecal administration of scAAV9-SMN1 is currently being evaluated in clinical trials in SMA type 2 patients (NCT03381729).
Amyotrophic lateral sclerosis (ALS) is characterized by motor neuron loss in multiple regions in the CNS.191,192 Dominant SOD1 mutations are most common in familial ALS; CRISPR-Cas editing of mutant Sod1 using AAV9-SaCas9 improved locomotor functions and survival when administered intravenously or via intracerebroventricular injection.193,194 A CSF-administered cytidine base editor was able to disrupt mutant Sod1 in adult ALS transgenic mice and improve survival. 195
Localized administration of CRISPR-Cas therapies in the brain is also essential for neurodegenerative diseases, with neuronal loss in specific areas of the brain such as the striatum in HD, limiting potential off-target effects in other tissues. Intracranial delivery of AAV1-SaCas9 targeting the 5′ end of exon 1 reduced mutant HTT expression in the HD mouse model and improved motor function and benefit survival. 196
CRISPR-Cas gene-editing therapy was also demonstrated in common progressive neurodegenerative disorders such as AD. Intracranial delivery of Cas9 nanocomplex to disrupt beta-secretase 1 (Bace1) in AD mouse models reduced amyloid plaque accumulation and cognitive deficits. 7 Dominant familial AD caused by mutations in amyloid precursor protein (APP), the site of beta-secretase 1 cleavage, results in accumulation of amyloid plaques. AAV9-Cas9 treatment via hippocampus injection specifically disrupted the mutant APP gene, reducing amyloid-β (Aβ) levels. 197 However, a possible limitation of the current treatment strategy is the lack of widespread targeting, as neurons in many areas of the brains are progressively affected in AD. Cas expression in the CNS can be driven by neuron-specific promoters while leveraging AAV tropism to increase target cell specificity. 197
Immunogenicity of the payload and vehicle
One of the most critical barriers to successful therapeutic translation is immune interaction. Most significantly, there are concerns that the adaptive immune response could inhibit the efficacy of CRISPR-based gene therapies. This could occur through the action of NAbs against either the Cas effector protein or the delivery vehicle, or through cytotoxic T cell responses leading to specific killing of treated cells.
Antibodies toward AAV and CRISPR-Cas9 have been shown to neutralize and negate editing efficacy in mice with just one previous exposure to the therapeutics. 198 This is particularly pertinent for clinical application, because multiple studies have now identified pre-existing immunity in the human population toward the commonly used Cas9 orthologs from S. pyogenes and Staphylococcus aureus,199–202 understandably since both are species that frequently colonize the human microbiome and may cause disease. Identification of circulating antibodies reacting to Cas9 could pose a large hurdle toward RNPs, since these purified Cas9 complexes could be neutralized within the bloodstream. Other Cas-encoding DNA and RNA modalities might generally be less inhibited by this obstacle, since the Cas9 protein will be expressed intracellularly and remain less accessible to antibodies from the circulation.
A more substantial risk, however, is the potential for CTL to recognize Cas-expressing cells through the Cas-derived peptides presented at the cell surface via the MHC class I pathway. Given the appropriate costimulatory milieu generated by innate immune signaling in the CTL, the target cell, and the surrounding tissue, this can lead to the killing of target cells expressing the Cas proteins.
Two in vivo mouse studies have highlighted the potential for anti-Cas9 immunity to inhibit the efficacy of CRISPR-based drugs. In one, a single dose of AAV-delivered Cas9 was shown to be sufficient to generate anti-Cas9 T cells in mice and to induce their killing potential. 198 Another study showed that prestimulated anti-Cas9 cytotoxic T cells completely negated the therapeutic effect of Cas9 gene editing within 12 weeks. 203 Although no human studies have shown an inhibitory effect of the immune system as yet, Cas9-reactive T cells clearly exist in significant fractions of the population.199,201 The interaction of the immune system with CRISPR-based therapies needs further study, but preliminary data suggest that it could become an obstacle to in vivo CRISPR-Cas therapies. Transient immunosuppression or transient transgene expression or Cas engineering might be necessary to maintain patient safety and efficacy. 204
Animal models
Several mouse models harboring disease-causing mutations have been generated for the study of CRISPR-Cas mediated gene editing (Table 1). Knockout mouse models have been essential in the study of inherited genetic diseases, including those affecting the CNS.205,206 Similarly, transgenic models bearing sporadic mutations have been generated for AD and Parkinson's disease. 207 Mouse models enable testing of the editing strategy, but they often do not fully recapitulate the extent of the disease. Mdx mice, for example, have a much slower disease progression, shortened lifespan relative to DMD patients, exhibiting cardiomyopathy only in older mice. 208 Likewise, AD mice exhibit the main pathology of the disease—Aβ aggregation—but overt neurodegeneration is not captured, nor early cognitive and behavioral symptoms. 209 In addition, it is harder to extrapolate results of widespread AAV delivery in mice organs such as the brain to humans due to differences in distribution 186 and possibly the large disparity in size.
Overall, advanced human diseases are better recapitulated in large animal models. The DMD pig model, for example, embodies severe disease progression such as premature death and cardiomyopathy. 210 Large transgenic animal models of HD also display severe phenotypes and early death when compared with rodent models. 211 Larger species such as dogs have a more similar brain disease profile to humans, and they develop naturally occurring LSDs.212,213 Large species are also more suitable for surgical manipulations, especially for neurodegenerative diseases. 214
Complexity can arise in the choice of animal species when progressing from small to large animal studies. For instance, vector tropism can differ between animal models as shown in the successful transduction of AAV5 in the mouse brain but not in cats. 215 In addition, although AAV-PHP.B has an enhanced ability to cross the BBB and increase CNS transduction in mice, this was not replicated in NHPs. 216 Extending preclinical studies to larger animals has been essential in better understanding immune responses toward gene therapy. Canine models have been essential in uncovering possible severe immune response against muscle-directed transgene 217 and cellular response toward AAV2 and AAV6 capsid. 218 Importantly, large animal studies can also inform immunosuppressive regimen for better therapy outcomes. 219 Interestingly, naturally AAV-infected rhesus macaques were unable to induce similar T cell responses to AAV capsids on re-exposure during treatment regimen despite the close phylogenetic relationship to humans.220,221
Readout challenges
Designing animal studies to provide an early readout for efficacy and potency is challenging, as shown in the preclinical study of EDIT-101. Editas Medicine used both a disease mouse model and NHP to measure desired physiological response. Well-established tissues in the eye allow for specific isolation, such as the cultivation of the neural retina of adult cadavers into retinal explant cultures for demonstration of editing efficacy in mature human photoreceptors. 222 Neural retinas from the knock-in mouse model of LCA10 were isolated to assess dosing to achieve functional rescue for clinical outcome. However, as photoreceptors in mice are predominantly rods, quantitative assessment of the editing rates in foveal cones was validated in a primate model. Surrogate guide RNAs had to be designed for NHPs due to its genetic divergence between humans. Total productive editing of the therapy was subsequently extrapolated from the editing frequency of the tissue sample. This extrapolation, in some instances relying on phenotypic versus genotypic outcomes, becomes particularly critical as obtaining human tissues (e.g., retina) may not always be feasible.
On the other hand, diseases entailing systemic correction such as DMD as opposed to localized editing in the eye will require multi-site sampling to quantify total editing. One of the drawbacks when using surrogate guides would be how representative the editing results will be in determining the correct dose for therapeutic efficacy. Greater confidence can be drawn by comparing with an additional model as shown in this study. For instance, EDIT-101 in the humanized mouse model, when compared with surrogate guides in NHPs, showed similar editing rates in response to increasing doses.
In contrast, the manufacturing of autologous products will vary due to the material received from the donor, but the final product can be well characterized before administration. Stadtmauer et al. quantified unique CRISPR-Cas9 engineered T cells for the frequency of edits by using a PCR assay, assessing for cytotoxic effects against tumor cells with an in vitro potency assay. 5 Unlike in vivo targeting, the editing efficiency required to bring about clinical benefit was not established for the targets. Whole blood samples and biopsy of the bone marrow and tumor site in patients provided a direct postinfusion readout of the therapeutic product.
An additional readout challenge is the careful characterization of the off-target genomic and transcriptomic effects of CRISPR-based therapies. In recent years, several sensitive methodologies for assaying off-target effects have been developed.223–229 Even so, the assaying of clinically relevant off-target effects remains difficult because depending on the genomic context and cell type, some off-targets may be completely benign, whereas others could have serious consequences. This is a well-recognized issue in the field and many efforts to manage this are underway, such as engineering the CRISPR payload at both the protein and gRNA level and management of the window of active exposure to the functional RNP complex. For more in-depth reviews on this aspect of CRISPR-based therapies see Refs.230–233
Regulatory concerns and n-of-1 trials
CRISPR-based therapeutics face a host of unique regulatory challenges when compared with other therapeutic methods such as small-molecule drugs or antibodies that typically act at the protein level. There are two layers of information abstraction when moving from DNA to RNA to protein, and often this information may have important implications for therapeutic development.
One such example is the fact that even a single genetic disease caused by knockout of a single gene may have multiple underlying mutations in different patients. Often a single mutation, such as the deltaF508 in cystic fibrosis, the maternal allele deletion of chromosome 15q11-13 in Angelman syndrome, and the 1278insTATC mutation in the HEXA gene in Tay-Sachs disease, accounts for a majority of cases, but in each of these diseases, tens to hundreds of other mutations affecting the same genes also give rise to the disease. Correction of these mutations by CRISPR-Cas genome editing would likely require different gRNA constructs, or even different Cas effector modalities, even with the same delivery mechanism. These different gRNAs would naturally have different safety and efficacy profiles as a result of the varying functional activities and off-target effects of distinct gRNAs. Under standard regulatory frameworks, this would seem to require many individual therapies based on the same platform to treat the same disease to undergo their own arduous and expensive approval studies.
Oligonucleotide-based drugs, particularly antisense oligos (ASOs), although ripe with therapeutic potential, face a similar regulatory problem. To address this, companies such as Ionis Therapeutics have pioneered n-of-1 trials with personalized ASOs for patient-specific mutations, causing a variety of diseases including ALS, Batten disease, and cystic fibrosis.234–236 Despite the success of several of these trials, it is an arduous process for which the FDA is not streamlined. These trials to date have relied on right-to-try clauses, expanded access INDs, and perhaps most critically, heroic fundraising efforts on behalf of the patients. There remains no clear regulatory path for these kinds of hyper-personalized medicines to be tested at scale. However, due to the trailblazing of ASO therapies and the promise of CRISPR-based drugs, the FDA and other global regulatory agencies are acutely aware of this problem and may well deploy new guidelines to mitigate it.237,238
Clearly, safety and efficacy must be demonstrated, even for n-of-1 trials and personalized therapies. 239 However, the process by which that can be accomplished must be simplified and streamlined for drugs that have an established platform. Many aspects of safety testing can be ported between similar drugs that may use the same delivery and mechanism, but with different underlying nucleic acid targets. In addition, inexpensive, fast, and robust efficacy testing platforms must be developed, which fortunately is an area of significant research interest. Although the landscape of current drug development models is not designed for the dynamic, personalized potential of the future of therapeutic breakthroughs, including CRISPR-based drugs, deft regulatory work and new guidelines can address this problem and pave the way to unlock that potential.
Conclusions
We are currently witnessing the birth of a new therapeutic paradigm based on CRISPR-Cas to precisely target genetic and epigenetic disease in a powerful and previously inaccessible manner. Given the rapid development and adoption of the CRISPR-Cas system, the explosion of preclinical studies, and the progress in clinical trials in the past few years, the near future will likely see the approval and deployment of several CRISPR-based therapies targeting diverse pathologies, some previously untreatable, and likely encompassing multiple therapeutic and delivery modalities. Despite this exciting prognosis, significant challenges remain, especially precise and high-quantity delivery to target tissues, demonstrated safety at the level of both off-target effects and immune interactions, and scalable, affordable deployment of the drugs. The substantial research efforts mounted toward all of these issues have yielded rapid advancements on every front, but much more remains to be done.
Although first-generation gene therapy Zolgensma garnered the epithet of “most expensive drug ever” at more than $2.1 million, we do not believe this must be a lasting attribute of this class of drugs. Although aspects such as AAV manufacturing challenges, R&D costs, regulatory burdens, and pharmaceutical market economics are important drivers of high price tags, the pace of technical progress can rapidly alter what once seemed set in stone. The precision and deftness with which we can manipulate genomes using CRISPR systems was virtually unthinkable just 17 years ago when the Human Genome Project was completed, just one year after the acronym CRISPR was first proposed. 240 Now, we have a thousand dollar genome and a million dollar gene therapy. Given the explosion of research and development into CRISPR-based gene therapy and the expansion of its application from rare genetic disease to large-market ubiquitous conditions such as chronic pain, 241 cardiovascular disease,242,243 and Alzheimer's, 7 it is not entirely implausible nor unprecedented in the decades to come to hope for a few thousand dollar gene therapy.
Optimizing the translation of new technologies to rapidly enable disease treatment is of critical importance but is far from easy. Many therapeutic modalities offering huge benefits have been stymied by unforeseen or unappreciated setbacks on the path to approval, often taking decades to address. We have attempted here to outline the current state and translational path forward of CRISPR therapeutics, highlighting areas where additional research is required, or potential challenges may occur to help orient thinking and optimize resources to solve these challenges. Expediting the development of these therapies will require vigilance, insight, and cooperation among scientists of multiple backgrounds, pharmaceutical companies, and regulatory stakeholders to ensure that the potential of CRISPR-based drugs may be realized as efficiently and safely as possible.
Footnotes
Author Disclosure Statement
P.M. is a scientific co-founder of Navega Therapeutics, Boundless Biosciences, Seven Therapeutics, Engine Biosciences, and Shape Therapeutics. The terms of these arrangements have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. W.L.C. is a scientific co-founder of Seven Therapeutics.
Funding Information
This work was generously supported by NIH grants (R01HG009285, RO1CA222826, RO1GM123313) and by the Agency for Science, Technology, and Research under its Industrial Alignment Fund (Pre-Positioning) (H17/01/a0/012).