
Abstract
Even for the genetically accessible yeast Saccharomyces cerevisiae, the CRISPR-Cas DNA editing technology has strongly accelerated and facilitated strain construction. Several methods have been validated for fast and highly efficient single editing events, and diverse approaches for multiplex genome editing have been described in the literature by means of SpCas9 or FnCas12a endonucleases and their associated guide RNAs (gRNAs). The gRNAs used to guide the Cas endonuclease to the editing site are typically expressed from plasmids using native Pol II or Pol III RNA polymerases. These gRNA expression plasmids require laborious, time-consuming cloning steps, which hampers their implementation for academic and applied purposes. In this study, we explore the potential of expressing gRNA from linear DNA fragments using the T7 RNA polymerase (T7RNAP) for single and multiplex genome editing in Saccharomyces cerevisiae. Using FnCas12a, this work demonstrates that transforming short, linear DNA fragments encoding gRNAs in yeast strains expressing T7RNAP promotes highly efficient single and duplex DNA editing. These DNA fragments can be custom ordered, which makes this approach highly suitable for high-throughput strain construction. This work expands the CRISPR toolbox for large-scale strain construction programs in S. cerevisiae and promises to be relevant for other less genetically accessible yeast species.
Introduction
The bacterial-derivative CRISPR-Cas technology is nowadays the most commonly used tool for microbial genome engineering. For the eukaryotic model and industrial workhorse Saccharomyces cerevisiae, several CRISPR-based methodologies have been developed, aiming at a fast and efficient single editing event.1–4 Two class II bacterial endonucleases, Cas9 and Cas12a (also known as Cpf1), have been functionally characterized for DNA editing, ranging from point mutation to heterologous pathway integration.4–6 While diverse Cas9- and Cas12a-mediated approaches for multiplex genome editing have been described in the literature (reviewed in Adiego-Perez et al. 7 ), multiplex genome editing still requires substantial effort for the CRISPR tools to be built. The RNA molecules designed to guide the endonuclease toward the editing site (guide RNAS; gRNAs) are typically cloned in and expressed from plasmids. In most published works so far, multiplex editing relies on the parallel transformation of multiple plasmids carrying a single or two gRNAs. However, this approach is limited by the number of available marker-based plasmid backbones.4,8–12 More recently, several successful examples have shown that multiple gRNAs can be expressed from a single gRNA array, using different tricks to release the mature gRNAs.3,5,6,13–17 However, the complexity of these gRNA expression cassettes and their tailored sequence design may be difficult to synthesize and require laborious and time-consuming cloning steps, therefore hindering the workflow for strain construction. To date, few attempts have been developed to circumvent gRNA cloning for genome editing of microbes in general and of S. cerevisiae in particular (illustrated in Fig. 1).

Cloning-free approaches for CRISPR-Cas-aided DNA editing. Overview of methodologies based on delivery of linear DNA templates for gRNAs expression in Saccharomyces cerevisiae. In vitro sample preparations and in vivo events upon transformation are described. All gRNA expression cassettes include an RNA polymerase III (RNA Pol III) terminator. Number of polymerase chain reactions (PCR) are quoted. Features are depicted in the legend on the right-hand side of the figure.
The most straightforward cloning-free strategy would rely on the delivery of the gRNA in the form of a short, linear DNA fragment. Such short DNA fragments could easily be synthetized as oligonucleotides and delivered as a mixture in any desired gRNA combination for multiplex targeting of DNA sites. Such a cost-effective and versatile approach would be highly suited for high-throughput, multiplex genome engineering of strains. Transient expression of linear DNA carrying gRNA expression cassettes has been previously shown to enable Cas9-mediated DNA editing.9,18 However, these approaches systematically require a first in vitro step for the construction of vectors from which the linear DNA is produced by polymerase chain reaction (PCR) amplification (Fig. 1). In eukaryotes, gRNAs are transcribed either by RNA polymerase III (RNA Pol III) or by RNA Polymerase II (RNA Pol II) promoter, the latter being flanked by self-processing ribozymes or tRNAs that prevent unwanted processing of the gRNAs.16,17 A recent report has shown that functional gRNAs can also be transcribed in different yeasts by the RNA polymerase from bacteriophage T7 (T7RNAP) localized in the nucleus. 19 Delivered as plasmid DNA, the T7RNAP-transcribed gRNAs have been used to guide Cas9 for genome editing and dCas9 for transcriptional regulation.
The present work introduces the gEL DNA method—a novel, utterly cloning, and PCR-free genome editing tool based on the

Schematic overview of the gEL DNA approach: 1, in silico design and ordering of gDNA cassettes (87 bp) and repair DNA (120 bp) as oligos; 2, transformation with the double-stranded (ds) gDNA expression cassettes (2a), the ds repair DNA fragments (2b), and an empty, split plasmid carrying a marker of choice (2c); 3, expression of the gRNA by the T7RNAP; 4, targeted DNA editing by FnCas12a; 5, repair of the dsDNA break via homologous recombination using the repair DNA fragments.
Methods
Strains and cultivation conditions
All S. cerevisiae strains used in this study (Table 1) were derived from the CEN.PK background strain. 20 Yeast cells were grown at 30°C in shake flasks on a rotary shaker (200 rpm) or on agar plates (20 g/L). Complex medium contained 10 g/L yeast extract, 20 g/L peptone, and 20 g/L glucose (YPD). YPD was supplemented with nourseothricin (100 mg/L), geneticin (G418; 200 mg/L), or hygromycin B (200 mg/L) to select transformants. Minimal synthetic media were prepared as previously described. 21 SMD medium contained 5 g/L (NH4)2SO4, 3 g/L KH2PO4, 0.5 g/L MgSO4·7H2O, 1 mL/L a trace element solution, supplemented with 20 g/L glucose, and 1 mL/L vitamin solution. SMD-urea included 6.6 g/L K2SO4, 3.0 g/L KH2PO4, 0.5 g/L MgSO4·7H2O, 1 mL/L trace elements solution, supplemented with 20 g/L glucose, 1 mL/L vitamin solution, and 2.3 g/L CH4N2O. 22 Utilization of urea as a nitrogen source instead of ammonium prevents excessive acidification of the medium resulting from ammonium uptake, and thereby enables the culture pH to be maintained close to the initially set value. For selection of transformants carrying the amdS marker cassette, ammonium sulphate in SMD was substituted with 10 mM acetamide and 6.6 g/L K2SO4 (SM-Ac). 23 Plasmids were propagated in Escherichia coli XL1-Blue cells (Agilent Technologies, Santa Clara, CA) after growth in Lysogeny broth (10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl) liquid culture (180 rpm) or solid medium (20 g/L agar) supplemented with chloramphenicol (25 mg/L), spectinomycin (100 mg/L), or ampicillin (100 mg/L) at 37°C. When required, plasmids from yeasts isolates were removed according to described procedures. 4 All S. cerevisiae and E. coli stocks were prepared by aseptically adding 30% v/v glycerol to exponentially growing cultures. Aliquoted cell stocks were stored at −80°C.
Saccharomyces cerevisiae Strains Used in this Study
Integration site at 194944–195980 of chromosome X from Mikkelsen et al. 28
Molecular biology techniques
Yeast genomic DNA used for cloning purposes was isolated using the YeaStar genomic DNA kit (Zymo Research, Irvine, CA) according to the manufacturer's instructions. Diagnostic PCR was performed using DreamTaq DNA polymerase (Thermo Fisher Scientific, Waltham, MA). For cloning and sequencing purposes, PCR products were obtained using Phusion® High-Fidelity DNA Polymerase (Thermo Fisher Scientific). Primers were ordered as polyacrylamide gel or desalted purified oligonucleotides (Supplementary Table S1) from Sigma–Aldrich (St. Louis, MO). Annealed oligos were quantified with a broad range dsDNA kit using a Qubit spectrophotometer (Invitrogen, Carlsbad, CA). DNA fragments were separated by electrophoresis on 1% (w/v) or 2% (w/v) agarose gels, depending on the fragment size. PCR products were purified using a GenElute™ PCR Clean-Up Kit (Sigma–Aldrich), after restriction digestion of the PCR mixture with DpnI (Thermo Fisher Scientific) for removal of circular templates. When required, DNA fragments were excised from gel and purified using a Zymoclean™ Gel DNA Recovery Kit (Zymo Research). Plasmids were isolated from E. coli cultures using a Sigma GenElute™ Plasmid Miniprep Kit (Sigma–Aldrich).
Entry-vector plasmids construction
All plasmids used in this study are listed in Table 2.
List of Plasmids Used in this Study
“sgRNA” denotes single-guide RNA used by SpCas9, “cr” refers to crRNA for FnCas12a. The presence of an “s” following the crRNA indicates that a shorter spacer of 19 nt is used. Otherwise, the spacer size is 25 nt.
The pUD565 plasmid, 24 a green fluorescent protein dropout (GFPdo) entry vector compatible with yeast toolkit parts, 25 was ordered as synthetic gene from GeneArt (Thermo Fisher Scientific). GFPdo entry vectors for cloning of transcriptional unit were constructed following the BsaI Golden Gate reaction protocol described by Lee et al. 25 The GFPdo pGGKd018 plasmid was obtained by assembly of part plasmids pYTK002, pYTK047, pYTK067, pYTK077, pYTK082, and pYTK085. The GFPdo pGGKd034 plasmid was constructed by assembly of part plasmids pYTK002, pYTK047, pYTK067, pYTK079, pYTK082, and pYTK083. The GFPdo pUDE810, an entry vector for FnCas12a-crRNAs, was constructed by Golden Gate assembly of pre-annealed primers 12647–12648 with the following PCR generated fragments: the pGGKd018 backbone with primers 12799–12800; the SNR52 promoter amplified from the pMEL13 template 4 using primers 12645–13546; and the GFPdo cassette bearing specific overhangs (GATC and ATCC) obtained by PCR amplification of primers 13547–12644 on pYTK047. 25
Construction of the dual Cas-expressing strain IMX1752
The construct for genomic integration of the Spcas9 gene consisted of a paired expression cassette for the introduction of Streptococcus pyogenes Cas9 nuclease and natNT2 marker into the SGA1 locus. 4 First, the natNT2 marker was PCR-amplified from pUG-natNT2 (Addgene plasmid #11092226) using primers 10297 and 10298. This PCR product was cloned via Golden Gate together with pre-annealed primer pairs 10293–10294 and 10295–10296, and yeast toolkit plasmids pYTK013, pYTK036, pYTK051, pYTK082, and pYTK083, 2 resulting in plasmid pUDE483. The SpCas9-natNT2 integration cassette was obtained by enzyme restriction of pUDE483 using EcoRI. The restriction mix was directly transformed into S. cerevisiae using the lithium acetate (LiAc) transformation protocol. 27 Transformants were selected on YPD supplemented with nourseothricin. A single isolate, which was renamed IMX1714 (Table 1), was submitted to an additional transformation for the genomic integration of the FnCas12a nuclease. For this, the sequence of Francisella novicida Cas12a was amplified from pUDC175 (Addgene plasmid #103019 5 ) using primers 13553–13554. The obtained PCR product, carrying 60 bp homology flanks to the X-2 integration site, 28 was transformed in IMX1714 as previously described in Gietz and Schiestl, 27 together with plasmid pUDR57329 for SpCas9-mediated targeting at this genomic site. Transformants were selected on SM-Ac plates. Correct genomic integrations were confirmed by diagnostic PCR using primers listed in Supplementary Table S1. After removal of the gRNA expression plasmid, the dual Cas9–Cas12a S. cerevisiae strain was stocked as IMX1752.
Construction of the T7RNAPK276R-expressing strain IMX1905
First, the T7RNAPK276R sequence was PCR-amplified from plasmid pRS315-nls-T7-RNAP (Addgene plasmid #33152) 30 using primers 13543 and 13544, and the obtained PCR fragment was stably cloned into entry vector pUD565, resulting in part plasmid pGGKp172. The T7RNAPK276R transcriptional unit was assembled by Golden Gate cloning into plasmid pGGKd034, together with part plasmids pGGKp035 (TDH3p) and pGGKp039 (TEF1t), 24 leading to plasmid pUDE866.
For genomic integration of the T7RNAPK276R, the previously characterized YPRCτ3 site of S. cerevisiae genome was chosen as the recipient locus. 31 Thus, a gRNA for FnCas12a-mediated editing at this site was designed according to guidelines provided in Swiat et al. 5 The gRNA for integration in YPRCτ3 was ordered as oligos 14142–14143 containing specific overhangs for Golden Gate assembly (GATC and ATCC). Oligo annealing and cloning into pUDE810 plasmid resulted in the crRNA-expressing plasmid pUDR477. Amplification of the T7RNAPK276R integrative cassette was carried out on pUDE866 plasmid using primers 14022 and 14023, which contain repair ends of 60 bp homologous to the YPRCτ3 locus. These generated PCR fragments were co-transformed with plasmid pUDR477 into IMX1752 cells, as previously described. 27 Yeast cells were selected on solid YPD plates supplemented with G418. Diagnostic PCR was performed on a single colony isolate, the plasmid was recycled, and the constructed strain was renamed IMX1905.
Construction of T7RNAP mutants and T7RNAP-overexpressing strains
In order to alter the T7RNAP protein sequence, the T7RNAP gene of IMX1905 was in vivo mutated by means of the CRISPR-SpCas9 editing machinery. A single gRNA was chosen for targeting the sequence surrounding DNA encoding amino acids at positions 266 and 276 (corresponding to 276 and 286 if considering the NLS; Supplementary Fig. S3). For this, oligo 14284 was Gibson assembled by bridging to the pMEL134 backbone, which was previously PCR amplified using primers 6005 and 6006. The obtained plasmid was renamed pUDR506. Repair oligos (Supplementary Table S1) consist of 120 bp surrounding the T7RNAP targeted sequence with single nucleotide polymorphism (SNPs) for P266L and/or R276K mutations and carrying a silent mutation at the protospacer adjacent motif sequence to avoid reiterative cutting. Plasmid pUDR506 and each of the double-stranded repair oligos were co-transformed into competent IMX1905 cells. 27 Transformants were plated on YPD agar supplemented with G418. Screening of eight selected colonies was performed by SNP genotyping with primers listed in Supplementary Table S1, following previously described procedures for SNP scoring. 32 After SNPs validation and Sanger sequencing of the mutated T7RNAP sequence (Supplementary Fig. S3), strains were stocked as follows: IMX2030 (T7RNAPP266L,K276R) was renamed after the P266L amino acid substitution; IMX2031 (wtT7RNAP) expresses the wild-type T7RNAP, where the arginine at position 276 is changed into the native lysine; and IMX2032 (T7RNAPP266L) resulted from simultaneous mutations of proline and arginine at positions 266 and 277 for the respective amino acid change in leucine and lysine.
For T7RNAPK276R overexpression, the dual Cas-expressing strain IMX1752 was transformed with plasmid pUDE866, following standard practice. 27 Transformants were selected on YPD plates supplemented with hygromycin B, and the strain was renamed as IME459. In parallel, transformation of IMX1752 with the empty vector pGGKd034 led to the control strain IME460. To overexpress the T7RNAPP266L variant, the gene sequence of strain IMX2032 was PCR amplified from its isolated genomic DNA using primers 10753 and 10768. The obtained PCR product was cloned by Golden Gate assembly into the episomal entry plasmid pGGKd034. The obtained plasmid, renamed pUDE911, was therefore transformed into IMX1752. Transformants were plated on selective YPD hygromycin B medium, and selected colonies were stocked as IME475.
Construction of the FnCas12a-T7RNAPP226L transportable system
To construct plasmids carrying FnCas12a expression cassettes, two alternative promoters were cloned by Golden Gate into the pGGKd034 vector together with the PCR product obtained by amplifying pUDC175 using primers 18075–18076. Plasmid pGGKp100 (PFK1p) 33 was used for assembly of pUDE1082, while plasmid pYTK027 (REV1p) 25 was used for the construction of pUDE1086. Cloning of the transportable FnCas12a-T7RNAPP226L system was performed by Gibson assembly using PCR amplicons with synthetic flags of 60 bp. Flagged primers 18077–18166 were used for PCR linearization of plasmid pUDE911. Flagged FnCas12a expression cassettes were amplified from pUDC175, pUDE1082, or pUDE1086 templates using primers 11868–10189, 18078–10189, or 18132–10189, respectively. Gibson assembly of these alternative FnCas12a expression cassettes to the linearized pUDE911 resulted in assembly of the respective plasmids pUDE1083, pUDE1084, and pUDE1087. CEN.PK113-7D transformed with each of the episomal plasmid were stocked as strains IME638 (pUDE911), IME639 (pUDE1082), IME640 (pUDE1083), IME641 (pUDE1084), IME642 (pGGKd034), IME645 (pUDE1086), and IME646 (pUDE1087). All transformants were selected on selective YPD hygromycin B medium.
Assembly of gRNA expression cassettes
The gRNA cassettes for evaluation of ADE2 deletion efficiencies mediated by FnCas12a or SpCas9 nucleases were prepared using the highly efficient ADE2-3 5 or the ADE2.y 1 gRNAs, respectively. Each gRNA cassette was expressed from high-copy plasmid and comprised the gRNA sequence left flanked by the RNA Pol III-dependent SNR52p, the minimal T7p TAATACGACTCACTATA (S.T7p) or an extended T7p GCCGGGAATTTAATACGACTCACTATA (L.T7p), with respective terminator sequences at the right flank. For the FnCas12a-mediated targeting of other genes, previously characterized gRNAs were expressed as single gRNA-expressing cassette or as in an array-like arrangement: HIS4 (HIS4-4), PDR12 (PDR12-3), or CAN1 (CAN1-4 or CAN1-3). 5
All single gRNA-expressing plasmids were assembled by Gibson assembly reaction using the NEBuilder® HiFi DNA Assembly Master Mix (New England Biolabs, Ipswich, MA). Depending on the plasmid features, the backbone of the pUDE810 plasmid was amplified using different primer couples for specific homology overhangs. The backbone for assembly of FnCas12a-gRNAs with SNR52p/SUP4t flanks was obtained by PCR amplification with primers 12710–5793. For T7RNAP-mediated expression of gRNAs via FnCas12a, plasmid backbone was obtained by PCR amplification using either primers 14274–13713 (S.T7p/T7t) or 14275–13713 (L.T7p/T7t). Plasmid pUDE759 was assembled by SNR52p/SUP4t backbone fragment with annealed oligos 12713–12714. Single oligos were used for the Gibson assembly of FnCas12a-gRNAs cassettes by single-stranded DNA bridging to each individual PCR-originated backbone fragments: the SNR52p/SUP4t derivatives pUDR482 (primer 14282), pUDR483 (primer 13750), pUDR484 (primer 14283), pUDR715 (primer 17328), pUDR716 (primer 17329), pUDR717 (primer 17330), and pUDR718 (primer 17331); the S.T7p/T7t-related plasmids pUDR485 (primer 14280), pUDR486 (primer 13751), pUDR487 (primer 14281), and pUDR488 (primer 13572); and the L.T7p/T7t cognate plasmids pUDR489 (primer 14276), pUDR490 (primer 14277), pUDR491 (primer 14278), and pUDR492 (primer 14279).
For assembly of SpCas9 gRNAs under SNR52p, the amplified pUDE810 backbone with SNR52p/SUP4t edges was mixed with annealed oligos 15508–15509 and the single-stranded oligo 14426 in a Gibson reaction, resulting in plasmid pUDR585. For SpCas9 gRNAs expressed by T7RNAP, T7-edged plasmid backbones were PCR amplified using primers 14274–15287 (S.T7p/sgRNA-T7t) or 14275–15287 (L.T7p/sgRNA-T7t), generating a dsDNA fragment that additionally contains a partial sequence of the gRNA scaffold for SpCas9. Gibson assemblies of annealed oligos 15290–15291 to either the S.T7p/sgRNA-T7t or S.T7p/sgRNA-T7t PCR-generated backbones were performed to obtain plasmids pUDR579 and pUDR581, respectively.
The control array of gRNAs expressed by plasmid pUDR692 was ordered as synthetic gene from GeneArt (Thermo Fisher Scientific). SNR52 promoter and gRNA design principles previously elucidated were used. 5 The synthetic gRNA array was flanked by BsaI sites and assembled by Golden Gate cloning into pGGKd018.
Delivery methods of gRNA expression cassettes
Each gRNA expression cassette was transformed together with 1 μg double-stranded deletion repair (Supplementary Table S1) in exponentially growing S. cerevisiae cells (∼2 × 107 cells/mL), according to the LiAc transformation protocol. 27 Genome editing via in vitro assembly described in Table 3 was prepared by transforming 500 ng (∼150 fmol) of each gRNA expression plasmid. For genome editing achieved via in vivo plasmid assembly, two linear PCR fragments were delivered with the transformation mix: (1) 150 fmol of the specific gRNA cassette, systematically amplified from the respective in vitro constructed plasmid using primers 14584–14585; and (2) 150 fmol of the linearized marked 2μ backbone with 60 bp homology to each gRNA cassette, obtained from the amplification of pUDE810 with primers 11571–12378. To evaluate genome editing via delivery of linear gRNA expression cassette, each amplified gRNA cassette (150 fmol) was transformed with either 500 ng circular pGGKd018 plasmid or with equimolar amount of two PCR fragments for the split plasmid selection using pGGKd018. These amplicons, having homologies for in vivo recircularization of the plasmid, were obtained by PCR amplification with primers 6815–9340 and primers 2398–12097.
Comparing gRNA Delivery Methods

SpCas9- and FnCas12a-mediated DNA editing efficiency in IMX1905 (Table 1) transformed with different delivery methods for gRNA-expression cassette.
All transformations were plated on selective YPD medium supplemented with G418. Efficiency of ADE2 deletion is measured as number of red colonies on total colony-forming units (CFU). For editing of other sites, diagnostic PCR was performed on a number of selected colonies using the primers listed in Supplementary Table S1.
Preparation of gDNAs and genome editing via gEL DNA
Sequences of deletion repair fragments and gDNAs are listed as forward and reverse oligonucleotides in Supplementary Table S1. Site XI-3 is located at the previously characterized integration locus on chromosome XI, 28 while XVI-1 corresponds to the intergenic region between CUP9 and TRE1 on chromosome XVI. The spacer for the non-targeting (nt) gDNA was designed by scrambling the ADE2-3s spacer sequence. Each forward and reverse oligo was mixed in equimolar amounts, heated for 5 min at 95°C, and cooled to room temperature. As the only exception, SpCas9-mediated editing using gDNA with long T7 promoter was obtained by PCR amplification of two overlapping primers: the gRNA-specific forward for ADE2.y (16745) and the universal reverse carrying the SpCas9-gRNA scaffold (16746). Concentrations of each double-stranded annealed oligos were measured for all pre-annealed oligos or the non-purified PCR-derived gDNA. One microgram of each deletion repair and 4 μg respective gDNA were mixed to 500 ng split pGGKd18 plasmid for selection purposes and transformed into competent T7RNAP-expressing yeast cells according to standard procedures. 27 Transformants were selected on YPD plates supplemented with G418 if transforming T7RNAP genomically integrated strains (IMX1905, IMX2030, IMX2031, IMX2032), or with G418 and hygromycin B if transforming T7RNAP-overexpressing strains (IME459, IME475) or for selection of the dual FnCas12a-T7RNAP overexpression strain (IME641). Plasmid-base controls for multiplex via FnCas12a gRNA arrays were performed according to Swiat et al. 5 Diagnostic PCR of selected colonies was done using the primers listed in Supplementary Table S1.
Growth rate measurement
Strains were cultivated on 96-well plates containing SMD medium or SMD-urea supplemented with hygromycin B (30°C, 250 rpm). Growth was monitored by measuring optical density at 660 nm at regular time intervals using the Growth Profiler 960 (Enzyscreen BV, Heemstede, Netherlands). Maximum specific growth rates (μmax) were calculated using the equation X = X0 eμt in which μ indicates the exponential growth rate from four independent biological cultures.
Bioinformatic analysis
The short sequence of the T7 promoter was mapped to CEN.PK113-7D reference 34 by using Bowtie aligner (v1.2.1.1), 35 with “–all” for reporting all alignments per input query.
The RNA secondary structure was predicted with the RNAstructure Web Server (https://rna.urmc.rochester.edu/RNAstructureWeb/). 36 The temperature was set to 30°C (303.15 K). Self-folding free energy is obtained via the same webtool.
Results
T7RNAP-expressing S. cerevisiae as a platform strain for Cas-mediated genome editing
For T7RNAP-based expression of gRNAs, the bacteriophage T7RNAP, previously functionally expressed in the yeast nucleus, was chosen. 30 Flanked by the strong and constitutive TDH3 promoter and TEF1 terminator, T7RNAP was integrated in the genome of a S. cerevisiae strain from the CEN.PK family that constitutively expressed both SpCas9 and FnCas12a (strain IMX1752; Table 1). Sanger sequencing of the resulting strain IMX1905 revealed a missense mutation in the coding sequence of the T7RNAP compared to the canonical sequence (https://www.uniprot.org/uniprot/P00573), which resulted in the replacement of a lysine by an arginine at amino acid 276 of the polymerase (corresponding to amino acid 286 when considering the NLS; Supplementary Fig. S1). As the strain characterized by Dower and Rosbash 30 contained the same amino acid substitution and was proven to be functional in yeast, we decided to keep this variant (from now on referred to as T7RNAPK276R) to test the gEL DNA approach.
Physiological characterization revealed that IMX1905, co-expressing T7RNAPK276R, SpCas9, and FnCas12a, grew as fast as the prototrophic control strain CEN.PK113-7D in chemically defined medium supplemented with glucose as sole carbon source (specific growth rate of 0.42 ± 0.01/h for IMX1905 and 0.44 ± 0.01/h for CEN.PK113-7D; Fig. 3). Expression of T7RNAP is therefore not toxic for S. cerevisiae.
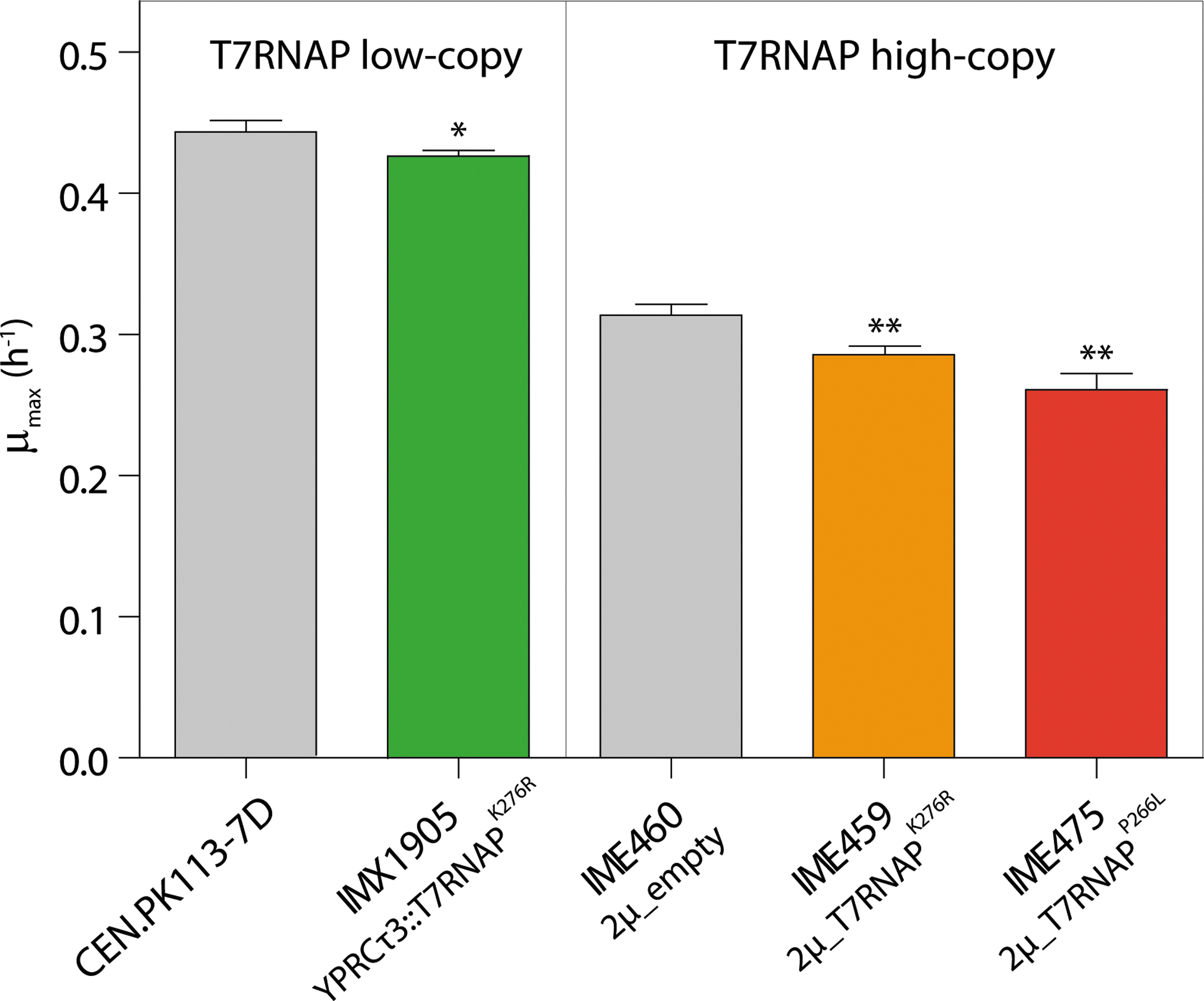
Physiological characterization of S. cerevisiae strains expressing T7RNAP. Maximum specific growth rates (μmax) of S. cerevisiae constitutively expressing T7RNAPK276R, SpCas9, and FnCas12a (IMX1905) and its control strain (CENPK.113-7D), or S. cerevisiae strains overexpressing T7RNAPK276R (IME459) or T7RNAPP266L (IME475) and its control strain carrying a 2 μm multi-copy empty vector (IME460). All strains were cultivated on a 96-well plate containing chemically defined medium supplemented with glucose as the sole carbon source (SMD for CENPK.113-7D or IMX1905; SMD-urea with hygromycin B for IME459, IME475, or IME460; the slower growth rate measured for strains with plasmids compared to strains with genomic integration is explained by the difference in medium composition). Data points represent average and mean deviations of four biological replicates. *p < 0.025; **p < 0.001. Student's t-test was calculated compared to respective control strains CENPK.113-7D or IME460.
T7RNAP enables gRNA expression from linear and circular DNA and promotes SpCas9- and FnCas12a-mediated DNA editing in S. cerevisiae
The activity of the T7RNAPK276R in S. cerevisiae was evaluated by measuring the DNA editing efficiency of SpCas9 and FnCas12a guided by gRNAs expressed from T7 promoter. Two different T7 promoter lengths were tested: the minimal T7 promoter of 17 bp (TAATACGACTCACTATA; referred to as S.T7p), and an extended T7 promoter sequence of 27 bp (GCCGGGAATTTAATACGACTCACTATA; referred to as L.T7p) known to improve the stability of the T7RNAP-promoter complex in vitro. 37 An in-depth alignment search showed that the S.T7p sequence does not occur in the S. cerevisiae genome, making unwanted transcription from the host genome highly unlikely. T7RNAPK276R-driven gRNA expression was compared to gRNA expression from the RNA Pol III–dependent SNR52 promoter, largely adopted by the yeast community for CRISPR-Cas editing.1,5 Downstream all three promoters, at the initially transcribed region, a guanine triplet was added to increase the T7RNAP transcriptional activity. 38 As previously reported for SpCas9, 19 the addition of this guanine triplet strongly improved T7RNAP-mediated expression for FnCas12a-based DNA editing (Supplementary Fig. S2). Disruption of ADE2, leading to a red colony phenotype, was used to assess editing efficiency 39 in strain IMX1905 (Table 1). Spacers previously shown to guide SpCas9 and FnCas12a to ADE2 with high efficiency were chosen.1,5 SpCas9 and FnCas12a have different requirements for functionality, which results in different compositions and size of the DNA cassette encoding the gRNA (from now on called gRNA cassette; Table 3). The ADE2.y gRNA for SpCas9 was used in its standard chimeric form, including the trans-acting RNA (tracrRNA). 1 The ADE2-3 gRNA for FnCas12a was reduced to the minimal 19 nt-long spacer enclosed by the matured direct repeats (DR), as recently described. 40 Thanks to FnCas12a minimal requirements for DNA targeting and editing (no tracrRNA, small DR and spacer), the gRNA cassettes for FnCas12a were substantially smaller than those for SpCas9 (Table 3).
The most popular method for gRNA delivery is via in vitro assembly of the gRNA expression cassette on a plasmid and transformation of this circular plasmid to yeast. As expected, expression of the gRNA cassette from SNR52p using this delivery method led to high efficiency in editing of the ADE2 gene in all colonies tested with both SpCas9 and FnCas12a (Table 3A). Conversely, T7RNAPK276R-based gRNA expression resulted in extremely low editing efficiency with FnCas12a (4.6 ± 0.2% for the short and 8.2 ± 3.2% for the long T7 promoter) and null or negligible editing with SpCas9 (Table 3A). Next to delivering a ready-made gRNA plasmid, two parts—one carrying the gRNA and the other the selection marker—were transformed into yeast. These two parts were flanked by 60 bp homologous sequences to enable in vivo circularization upon transformation. This delivery method enabled the transient availability of the gRNA cassette as linear DNA fragment. While reducing editing efficiency for SNR52p-based gRNA expression by around 10%, this method slightly increased ADE2 editing by T7RNAPK276R-mediated expression of gRNAs for both SpCas9 and FnCas12a (Table 3B). Editing by SpCas9 remained extremely low (around 1%), while up to 20% of the colonies displayed the disruption of ADE2 by FnCas12a (Table 3B). Next, to test whether the gRNA could be solely expressed from a linear DNA molecule, the gRNA cassette was delivered as double-stranded DNA fragment. A plasmid carrying a selectable marker was transformed in parallel for selection purposes. With this delivery method, editing efficiency with RNA Pol III–mediated gRNA expression was dramatically reduced to around 10% with both SpCas9 and FnCas12a (Table 3C). Editing efficiency for T7RNAPK276R-based gRNA expression was also reduced but still detectable at around 6% when using FnCas12a. It has been shown that the efficiency of SpCas9-mediated DNA editing can be increased by supplying a split plasmid during transformation, presumably by offering a selective advantage to cells that are proficient in homology directed repair. 9 Accordingly, a twofold increase in ADE2 editing efficiency was measured with both SpCas9 and FnCas12a when the gRNAs were transcribed by RNA Pol III (Table 3C and D). However, using a circular or a split plasmid did not affect DNA editing for T7RNAPK276R-expressed gRNAs (Table 3C and D). The split marker approach combined with linear DNA delivery of the gRNA described in Table 3D was nonetheless kept for all the following experiments.
Altogether, these data demonstrate that gRNAs can be expressed from circular and linear DNA using the T7RNAK276R polymerase in S. cerevisiae. Additionally, FnCas12a leads to higher DNA editing efficiency than SpCas9 when guided by T7RNAPK276R-expressed gRNAs. In all experiments, the long T7 promoter consistently led to two- to threefold higher editing efficiencies than the short T7 promoter (Table 3A and B) as a possible consequence of the higher T7RNAP-promoter complex stability. 37 While delivery of gRNAs in the form of short linear DNA fragments enabled DNA editing, the observed editing efficiencies were low and required further optimization to turn the gEL DNA approach into an attractive and competitive DNA editing technique.
Improving the efficiency of the T7RNAPK276R-based gEL DNA technique by optimizing the gDNA design
Aiming for cloning-free genome editing, the gEL DNA technique relies on the simple utilization of customized double-stranded DNA oligos (referred to as gDNAs) for the in vivo T7RNAPK276R-mediated expression of gRNAs. To reduce synthesis costs and increase compatibility with high-throughput strain construction, the size of the gDNA should be as small as possible and should not exceed 120 nt—the standard size limit of commercial custom-made oligomers. In this respect, FnCas12a presents a clear advantage, as its gRNAs consists of a smaller structural part (DR) than the one required for DNA targeting by SpCas9 (gRNA scaffold). Consequently, the small size of FnCas12a gRNAs gives more flexibility in gDNA design regarding the length of T7 transcriptional elements and the presence of terminal DR or T7 terminator. Conversely, a minimal gDNA configuration for SpCas9-mediated editing containing the S.T7p and the chimeric gRNA is 119 nt long (Fig. 4), which does not leave room for additional potentially useful parts such as a longer T7 promoter or a T7 terminator. As previously, a triplet of guanine was appended to the T7 promoter for all tested gDNA.
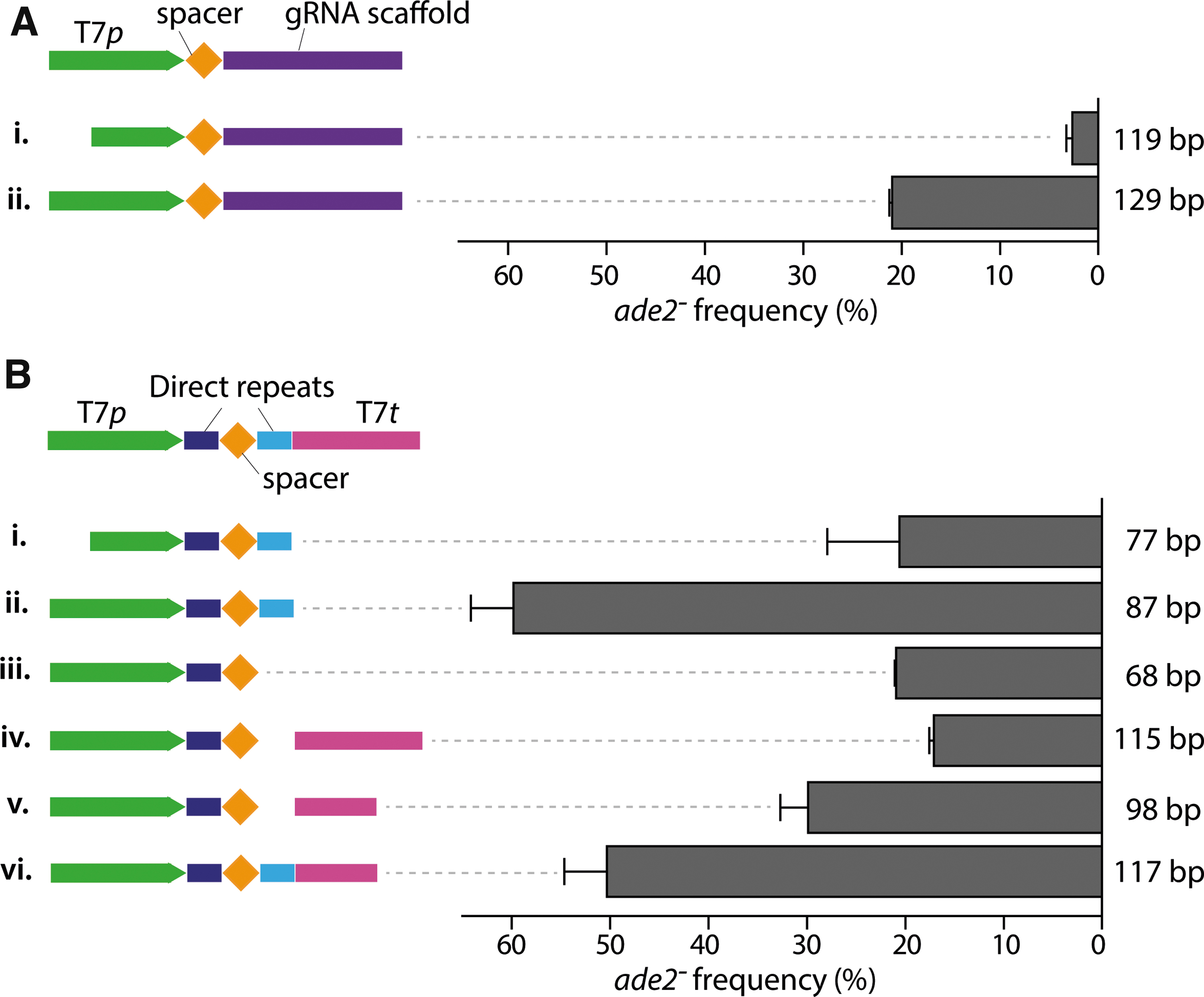
Optimization of SpCas9 and FnCas12a gDNA design. Editing efficiency of ADE2 in strain IMX1905 transformed with gDNAs for cloning-free, T7RNAP-driven expression of gRNA.
For SpCas9, two gDNA configurations were tested: one with the short and one with the long T7 promoter, followed by the ADE2.y spacer and the gRNA scaffold (Fig. 4A). Compared to the design presented in Table 3, no T7 terminator was added at the end of the gDNA. While the pre-annealed S.T7p gDNA can be directly transformed into IMX1905, the longer L.T7p gDNA (129 bp) had to be obtained by a preliminary PCR using two primers with overlapping homologies (see Methods). Both gEL DNA configurations did enable SpCas9-mediated DNA editing, marginal for the S.T7p (2.6% editing efficiency; Fig. 4A, i) but substantial for the L.T7p (21% editing efficiency; Fig. 4A, ii).
Six different gDNA configurations for the ADE2-3 target were tested for FnCas12a-mediated editing, differing in the size of T7 promoter and terminator as well as in the addition of a terminal DR and a T7 terminator (Fig. 4B). After simple pre-annealing of two complementary oligos in vitro, each gDNA was directly transformed into strain IMX1905. These data revealed that the terminal DR is important for efficient editing of ADE2, irrespective of the presence of the T7 terminator, and that the presence of a T7 terminator is not required (Fig. 4B). These results also further confirmed that the long version of the T7 promoter markedly increased DNA editing efficiency (Fig. 4B, i and ii). This design optimization enabled the DNA editing efficiency to be increased to 60%, relying on the very simple transformation of yeast with an 87 nt-long oligonucleotide. This simple and efficient design, represented in Figure 4B, ii, was implemented for the rest of the work with FnCas12a.
The editing efficiencies shown in Figure 4 were substantially higher for both SpCas9 and FnCas12a than those reported in Table 3D, in which a similar approach with linear gDNA delivery together with a split plasmid was also used. This increased efficiency most probably resulted from the higher amount of gDNA supplied to the transformation mix used for the experiments presented in Figure 4 (300- to 400-fold higher). The increased editing efficiency combined with the higher abundance of gDNA suggests that a greater supply of the gDNA to the cell might be a key element for genome editing using gEL DNA.
Improving the efficiency of the gEL DNA technique by optimizing sequence and expression levels of the T7RNAP
To explore further whether gDNA—and consequently gRNA availability—might be limiting editing efficiency, gDNA transcription efficiency by the T7RNAP was explored. To this end, three additional T7RNAP variants were tested. All three variants were constructed from IMX1905 by inserting point mutations in the T7RNAPK276R gene. In the first variant, the K276R mutation was reverted into the wild-type T7RNAP (wtT7RNAP, strain IMX2031). The second variant carried the P266L mutation, known to reduce abortive transcription in vitro 41 (T7RNAPP266L, strain IMX2032), and the third variant carried the two mutations (T7RNAPP266L, R276K, strain IMX2030). When tested with FnCas12a, all four variants enabled DNA editing with a significantly higher efficiency for T7RNAPP266L (Fig. 5), while T7RNAPK276R and T7RNAPP266L, R276K showed the lowest DNA editing efficiency (Fig. 5), suggesting that the K276R substitution is deleterious for T7RNAP transcription efficiency.
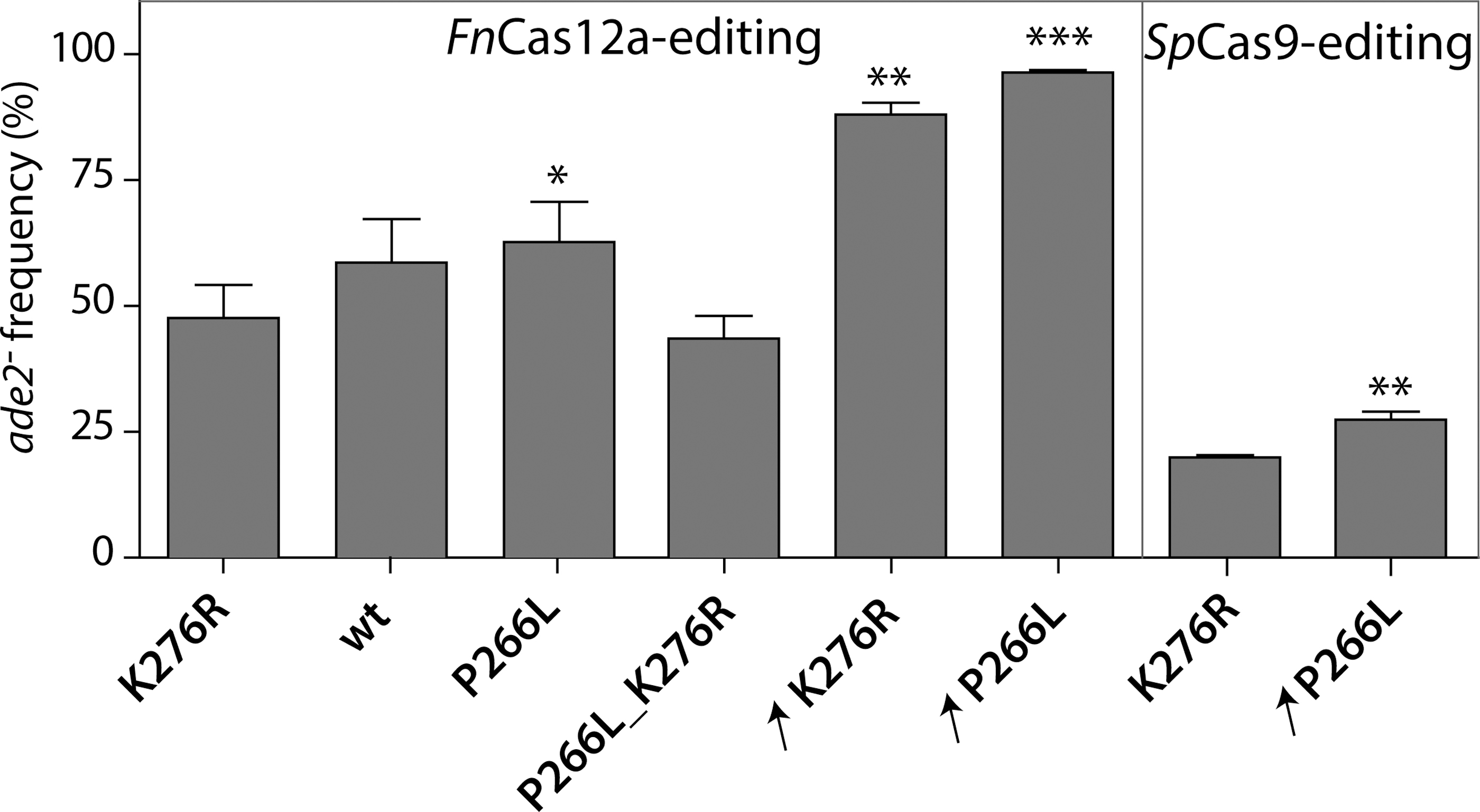
Comparison of SpCas9 and FnCas12a editing efficiency with T7RNAP variants. Efficiency of ADE2 editing by FnCas12a- or SpCas9-mediated gEL DNA in T7RNAP mutant or overexpression strains: IMX1905 (K276R); IMX2031 (wild-type, wt); IMX2032 (P266L); IMX2030 (P266L_K276R); IME459 (K276R overexpression, ↗K276R); and IME475 (P266L overexpression, ↗P266L). For FnCas12a, transformed gDNA corresponds to annealed 15093–15094 oligos. For SpCas9, transformed gDNA was obtained by PCR-derived fragment using overlapping primers 16745–16746. Editing efficiency is expressed as percentage of red colonies (ade2–). Values represent the average and standard deviations of data obtained from independent biological duplicates. *p < 0.05; **p < 0.025; ***p < 0.001. Student's t-test was calculated compared to respective control strain IMX1905 (K276R).
To enhance gDNA transcription efficiency further, the expression level of the T7RNAP was increased. The strains IME459 and IME475 were constructed by transformation with episomal plasmids harboring the T7RNAPR276K and the T7RNAPP266L variants, respectively. While expression of T7RNAP from a single integrated gene copy did not affect growth of S. cerevisiae (Fig. 3), expression from episomal vectors significantly reduced the growth rate when compared to a control strain carrying an empty episomal vector (0.29 ± 0.00/h for IME459, 0.26 ± 0.01/h for IME475, and 0.31 ± 0.01/h for the control strain IME460; Fig. 3 and Table 1). Overexpressing of either T7RNAP strongly increased the DNA editing efficiency by FnCas12a, approaching 100% when using T7RNAPP266L (Fig. 5). Overexpression of T7RNAPP266L also increased DNA editing efficiency by SpCas9 compared to a single copy of T7RNAPK276R, albeit it to a lesser extent (increase by 10%; Fig. 5). The gEL DNA approach remained much more efficient with FnCas12a than with SpCas9 (maximum efficiencies of 96% and 29%, respectively; Fig. 5). Finally, to test whether the efficiency of gEL DNA was sensitive to the location and sequence of the edited site, using these optimized conditions, three new sites were selected in non-coding regions and were tested for FnCas12a-mediated targeting with the T7RNAPP266L-overexpression strain (IME475). The high efficiency of single gEL DNA editing was confirmed with efficiency between 90% and 100% for these three loci (XI-3, 28 XVI-1, and YPRCτ3 loci; Supplementary Fig. S4).
Altogether, these results revealed that the expression levels of the T7RNAP, and therefore most likely gRNA availability, play a key role for successful DNA editing by FnCas12a in the gEL DNA system.
Assessment on the components required for efficient DNA editing using gEL DNA
In the approach described above, gEL DNA requires five components: the T7RNAP and FnCas12a already present in the transformed strain, and the gDNA cassette, repair DNA, and split plasmid carrying the selection marker, delivered as linear fragments during transformation. Control experiments in which these components were systematically omitted confirmed that all five components are required for efficient DNA editing using the gEL DNA method (Fig. 6 and Supplementary Fig. S5). Editing was completely abolished in the absence of targeting gDNA and of repair DNA, as well as if the targeting gDNA was replaced by a non-targeting variant, with or without repair DNA. The supply of the split plasmid is understandably not essential for DNA editing using the gEL DNA method but is key for the selection of edited transformants, as its absence resulted in an abundance of colonies on the plates and very low editing efficiency (around 8%). It is, however, remarkable that even in the absence of selection marker, correctly edited transformants could be found. It is interesting to note that DNA editing was observed in the absence of T7RNAP, albeit with extremely low efficiency (around 8%). This editing was, however, abolished when FnCas12a was also omitted. As the presence of targeting gDNA is necessary for DNA editing by FnCas12a (Fig. 6; no editing in the absence of targeting gDNA), this T7RNAP-independent editing might be explained by low-level transcription of the supplied gDNA by one of the native yeast polymerases or by guiding of FnCas12a by single-stranded gDNA present as contamination of the double-stranded gDNA stock, although both hypotheses seem unlikely. Overall, FnCas12a, T7RNAP, gDNA, repair DNA, and selection plasmid are essential for maximal DNA editing efficiency by gEL DNA.
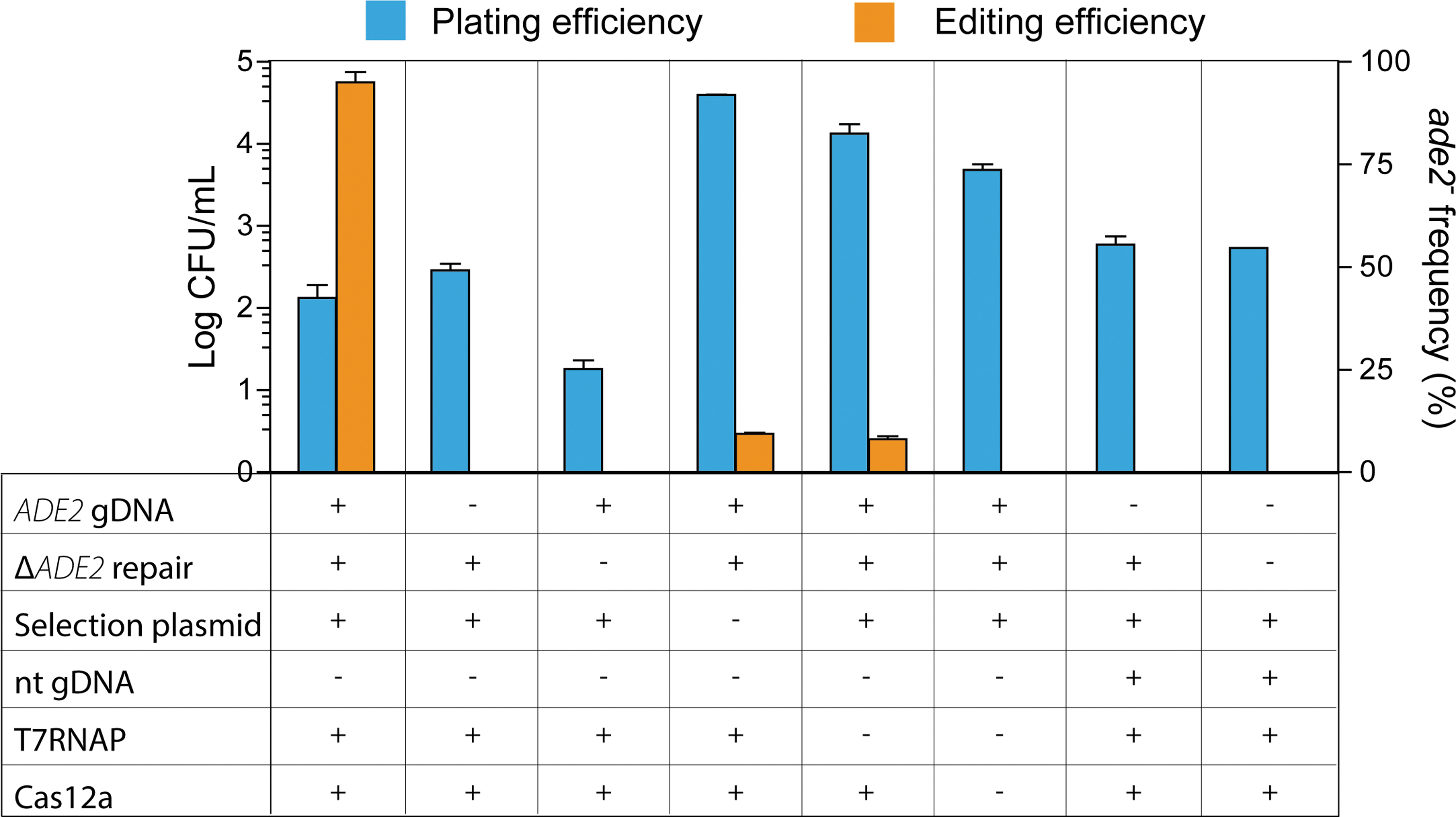
CFU and ADE2 editing efficiencies with different combinations of the components of the gEL DNA system. S. cerevisiae strains carrying FnCas12a and T7RNAP (IME475), FnCas12a alone (IMX1714), or control strain (CEN.PK-113.7D) were transformed with ADE2 or non-targeting (nt) gDNAs, alternatively omitting repair DNA for ADE2 deletion or split pGGKd018 plasmid for selection. Results of cell counts are presented as log CFU/mL. Editing efficiency is expressed as percentage of red colonies (ade2–), which were further verified by PCR (Supplementary Fig. S5).
gEL DNA enables FnCas12a-mediated multiplex genome editing in S. cerevisiae
To test for multiplex genome editing, four gRNAs targeting CAN1, HIS4, PDR12, and ADE2, previously shown to lead to 100% DNA editing efficiency by FnCas12a when expressed from an RNA Pol III promoter, were selected (Swiat et al. 5 ; Fig. 7A). As done for the ADE2-3 target used for singleplex gEL DNA, these additional gRNAs were shortened to a 19 bp-long spacer compared to the previously described plasmid-based constructs. 5 Oligos carrying the gDNA design shown in Figure 4B, ii, were ordered for each gRNA (Supplementary Table S1) and transformed in duplex or quadruplex to IME475 overexpressing the T7RNAPP266L. Duplex targeting of ADE2 and HIS4 revealed that the vast majority (14/16) of tested clones were edited and that 63% of the clones carried a double deletion (Fig. 7B). Out of the clones with single editing, none carried a single HIS4 deletion, while duplex editing with ADE2 was clearly a frequent event (Supplementary Fig. S6). Quadruplex targeting resulted in a substantial fraction of clones without any editing (34%; Fig. 7C). The fraction of clones with a single editing event was very similar for duplex and quadruplex editing (25% and 30%, respectively). Twenty-three percent and 13% of the clones carried double and triple editing, respectively, and quadruplex editing was not observed (Fig. 7C). Remarkably, none of the tested clones displayed editing in CAN1 (Supplementary Fig. S7), suggesting that the CAN1-4 gRNA failed to guide FnCas12a to the targeted site. This lack of targeting might be explained by the fact that the CAN1-4 gRNA contained an additional guanine triplet and was six nucleotides shorter than the CAN1-4 gRNA originally tested by Swiat et al. To test this hypothesis, the CAN1-4 gDNA (GGG at 5′ and a 19 bp-long spacer) was expressed from a plasmid with the SNR52 promoter and tested for editing efficiency. Out of eight selected colonies, none resulted in a CAN1 deletion (Supplementary Fig. S8)—a complete loss in editing efficiency that is likely due to the disruption of the gRNA stem-loop structure (Supplementary Table S3). A new CAN1 spacer with a predicted secondary structure displaying the gRNA stem loop was therefore selected for CAN1 targeting (CAN1-3 5 ; Supplementary Table S3). Expressed from a plasmid with the SNR52 promoter, CAN1-3 led to 100% CAN1 editing with FnCas12a (Supplementary Fig. S8). However, when tested for multiplexing using the gEL DNA methodology, CAN1-3 rarely led to editing of CAN1 by FnCas12a (Fig. 7D). A single CAN1 editing event was observed out of 30 clones tested (Supplementary Fig. S9), and remarkably, this event was concomitant with the editing of the three other targets, leading to a single clone with quadruple DNA editing (Fig. 7D). In the quadruplex editing experiments with CAN1-4 and CAN1-3, the fraction of clones with single, double, and triple DNA editing was comparable (roughly 30%, 25%, and 10%, respectively; Fig. 7C and D).
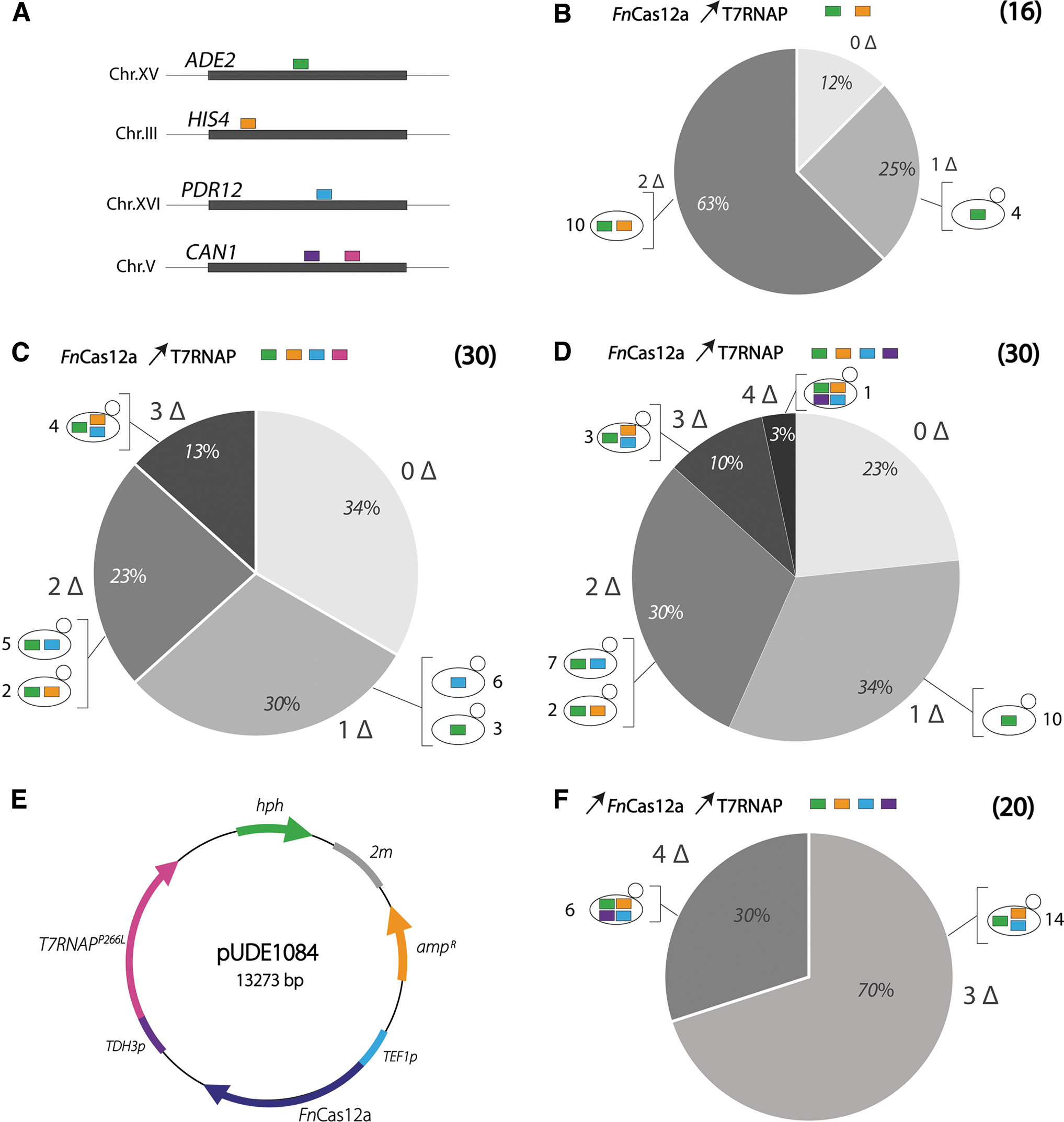
Multiplex genome editing by FnCas12a-mediated using the gEL DNA approach.
Following the approach described by Swiat et al., two cRNA arrays were tested for quadruplex genome editing. Both plasmids carried the HIS4-4, ADE2-3, and PDR12-3 gRNAs, but pUDE735 expressed CAN1-4 (Supplementary Fig. S7), while pUDR692 expressed CAN1-3 (Supplementary Fig. S9). As previously observed, the number of colonies obtained after transformation was extremely low (<10 colonies) compared to the number of colonies obtained for quadruplex editing with the gEL DNA approach (>150 colonies).
Construction and validation of a portable gEL DNA toolkit
The orthogonality of the T7RNAP-based gEL DNA system has great potential for other organisms. To demonstrate transportability, all-in-one multicopy plasmids carrying both T7RNAP and FnCas12a were constructed (pUDE1083, pUDE1084, and pUDE1087; Table 2). Both proteins have been shown to reduce growth rate when expressed individually at a high level from multicopy plasmids (this work for T7RNAP and Swiat et al. 5 for FnCas12a), simultaneous high-level expression of these two proteins might therefore be detrimental for the yeast strains. The results above show that the efficiency of DNA editing by gEL is sensitive to T7RNAP abundance. It was therefore decided to keep the same promoter for T7RNAP expression (TDH3p) but to tune the expression of FnCas12a by using three constitutive promoters spanning a broad range of strengths: REV1p with low expression (resulting in strain IME646), PFK1p with intermediate expression (strain IME640), and the strong TEF1p (strain IME641). As expected, co-expression of T7RNAP and FnCas12a decreased the specific growth rate compared to strains expressing T7RNAP or FnCas12a alone and to the control strain (around 20% decrease; Supplementary Fig. S10). However, strains with different promoter strengths for FnCas12a expression displayed similar growth rates (specific growth rate of 0.26 ± 0.01/h for IME640, 0.25 ± 0.01/h for IME641, and 0.25 ± 0.01/h for IME646; Supplementary Figure S10). In the absence of promoter-dependent phenotypic effect, the strain expressing FnCas12a under the control of the strongest, TEF1p promoter (IME641), was selected to test the DNA editing efficiency of the portable gEL DNA system (plasmid map at Fig. 7E). Singleplex editing of ADE2 revealed similar efficiencies between the integrated and portable systems (>95 ± 1% of edited colonies; Supplementary Fig. S11). However, the portable system proved to be substantially superior for duplex and quadruplex editing (Fig. 7F and Supplementary Figs S12 and S13), with 100% and 30% efficiency for duplex and quadruplex editing, respectively. Remarkably, the remaining 70% of the colonies transformed for quadruplex editing displayed triplex editing, with CAN1 systematically unedited (Fig. 7F). The plasmid-based gEL DNA approach is therefore highly efficient for singleplex and multiplex editing. It does not require a priori modification of target strain and is therefore a promising tool to be used in other yeasts, or even other organisms upon construction of compatible T7RNAP and FnCas12a-expressing plasmids.
Discussion
The future of the CRISPR-Cas-based genome editing is heading toward the development of fast and low-cost methodologies for strain construction. The gEL DNA approach presented in this study expands the CRISPR-Cas genome editing toolbox of S. cerevisiae with an entirely cloning-free and very efficient strategy for single or double genetic modification in S. cerevisiae. By simply transforming pre-annealed 87 nt-long complementary DNA oligonucleotides into competent yeast cells, the cost and time of strain construction can be reduced to the bare minimum. Any chosen gRNA cassette can be delivered independently or in combination with other gRNA cassettes, making this technique very versatile and highly suitable for high-throughput, combinatorial strain construction. Akin to other CRISPR-based techniques for genome engineering, increasing the number of simultaneously targeted sites strongly affects the efficiency of multiplexed gEL DNA or the viability of cells in terms of CFU on transformation plate (Supplementary Table S2 and Fig. 7).5,7 The results obtained in this study suggest that this efficiency can be further enhanced. For instance, increasing T7RNAP and gDNA abundance substantially increased singleplex gene editing (Figs 4 and 5), suggesting that gRNA abundance might be a key factor for efficient DNA editing. Measurement of gRNA abundance should be performed to confirm this hypothesis. However, measurement of these extremely short, transiently expressed RNAs during transformation is technically extremely challenging. While the toxicity of plasmid-borne T7RNAP expression showed that its abundance cannot be further increased in S. cerevisiae, the efficiency of the gEL DNA could be further enhanced by T7RNAP protein engineering or by expression of DNA-dependent RNAP variants from other bacteriophages (i.e., T3, SP6, or K11) that are able to transcribe from short promoters and from linear DNA templates.42–44 Another aspect to consider is the stability of the gDNA. While other methods deliver gRNA in the form of plasmids that are very stable in vivo, the linear nature of the gDNA makes it prone to degradation by native exonucleases. Further studies should explore the stability of gDNA and gRNA during transformation and test whether chemical stabilization of the linear gDNA (e.g., by phosphorothioate derivatives or 2′-ribose modification45,46) enhances gRNA availability and thereby DNA editing. There are therefore several promising avenues to improve multiplex DNA editing further with the gEL DNA approach.
Out of the eight gRNAs tested in this study, one failed to guide FnCas12a for gene editing. Remarkably, for this guide (CAN1-4), the folding prediction suggested the whole disruption of the DR as a consequence of the 5′ addition of the guanine triplet, while the other seven guides displayed typical gRNA secondary structures with the required stem-loop structure (Fig. 7 and Supplementary Table S3). 47 In agreement with these observations, a recent study about the FnCas12a gRNA functionality suggests that the disruption of the DR pseudoknot structure by pairing to the spacer sequence might lead to loss of gRNA targeting ability. 40 Additionally, inhibition of the gRNA processing and consequently of FnCas12a activity seems to be due to the positional effect of a stable secondary structure flanking the DR. 48 It has been recently advised that the terminator should be spaced out by a 24 nt-long spacer to avoid steric effects with the pseudoknot formation and thereby allow correct gRNA folding. 40 Our findings support these theories, since a gRNA flanked by the short 30 nt T7 terminator sequence that lacks the stem-loop structure has a 1.8-fold higher ADE2 editing efficiency than a gRNA with the longer 47 nt T7 terminator (Fig. 4B, iv and v). Prediction of the gRNA structure is therefore essential to optimize FnCas12a-based DNA editing with the gEL approach.
Despite efforts to improve editing with both SpCas9 and FnCas12a, the latter proved to be more efficient for DNA editing with the gEL DNA method. The causes for SpCas9's lower efficiency remain to be elucidated, but the observation that increasing T7RNAP abundance hardly affects DNA editing by SpCas9 (increased by 1.4%; Fig. 5) suggests that gRNA abundance is not the factor impairing SpCas9 activity. While the length of the gDNA might be another obstacle for SpCas9 implementation with the gEL DNA approach, it could be overcome by expressing the tracrRNA separately from the gRNA. 3
In conclusion, the gEL DNA methodology is not only an extremely valuable tool for genome editing in S. cerevisiae but has a yet greater potential thanks to its portability to other organisms. Expression of gRNAs using the host machinery or in vivo burden of gRNA expression plasmids can present serious obstacles for CRISPR-Cas9-based editing.2,49–52 By introducing a T7RNAP and gDNA oligos, the gEL DNA approach dissociates gRNA production from the host polymerase and from plasmid templates, thereby entirely removing these obstacles.
Footnotes
Acknowledgments
We thank Melanie Wijsman and Ewout Knibbe for constructing strains IMX1714 and IMX1752, respectively; Sofia Dashko for cloning plasmid pGGKd034; and Marcel van den Broek for the sequence alignment of the T7 promoter to the whole S. cerevisiae genome.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research received funding from the Netherland Organization of Scientific Research (NWO).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.