
Abstract
CRISPR guide RNAs (gRNAs) can be programmed with relative ease to allow the genetic editing of nearly any DNA or RNA sequence. Here, we propose novel molecular architectures to achieve RNA-dependent modulation of CRISPR activity in response to specific RNA molecules. We designed and tested, in both living Escherichia coli cells and cell-free assays for rapid prototyping, cis-repressed RNA-interacting guide RNA (igRNA) that switch to their active state only upon interaction with small RNA fragments or long RNA transcripts, including pathogen-derived mRNAs of medical relevance such as the human immunodeficiency virus infectivity factor. The proposed CRISPR-igRNAs are fully customizable and easily adaptable to the majority if not all the available CRISPR-Cas variants to modulate a variety of genetic functions in response to specific cellular conditions, providing orthogonal activation and increased specificity. We thereby foresee a large scope of application for therapeutic, diagnostic, and biotech applications in both prokaryotic and eukaryotic systems.
Introduction
The CRISPR loci and the associated Cas proteins constitute a type of prokaryotic adaptive immune system. 1 According to the type, CRISPR loci include a variable number of gene coding sequences, identified as CRISPR-associated (Cas) genes, linked to a repeat-spacer array by the leader sequence. The array contains short repeats separated by unique spacers acquired from invading viral RNAs or plasmids after the first infection (adaptation). These are transcribed and processed into small RNAs (sRNAs) that associate with one or more Cas proteins to form the active ribonucleoprotein (RNP) complex (expression). The RNA spacers are used to target specific nucleic acid sequences of mobile genetic elements upon subsequent invasions, activating the protein part of the RNP complex to cleave or degrade the invading DNA or RNA sequences (interference). In the most commonly exploited type II CRISPR system found in Streptococcus pyogenes, a single Cas9 protein acts as RNA-guided endonuclease by forming a complex with the CRISPR RNA (crRNA) and the trans-activating crRNA (tracrRNA) fragments to bind and cleave the double-stranded DNA target. 2 A single engineered RNA can also be used, by fusing the crRNA and the tracrRNA into one short synthetic guide RNA (identified as sgRNA or just gRNA). The gRNA is formed of three main domains: (1) the crRNA section, including the ∼20 nt targeting region (or spacer) and the 22 nt segment base pairing with the tracrRNA complementary region; (2) the tracrRNA region complementary to the crRNA; and (3) the 3′ handle region, which allows termination of transcription of the gRNA fragment as well as stabilization of the Cas9-gRNA RNP. After RNP formation, the targeting region of the crRNA (or sgRNA) remains single stranded, with the 12 nt of the targeting region proximal to the Cas9 handle (identified as “seed” sequence) maintained in an A-form conformation to allow its hybridization with the complementary DNA sequence targeted. 3 The Cas9 handle is composed of four stem loops, the first being formed by the interaction between crRNA and tracrRNA (linked through a short loop in the sgRNA) followed by three smaller stems, the nexus, and the two 3′ terminal hairpins (Fig. 1A). These secondary structures have proved crucial for the formation of an active Cas9-gRNA RNP, which can be affected if the targeting region contains significant secondary structures 4 or modifications are added to its 5′ end. 5
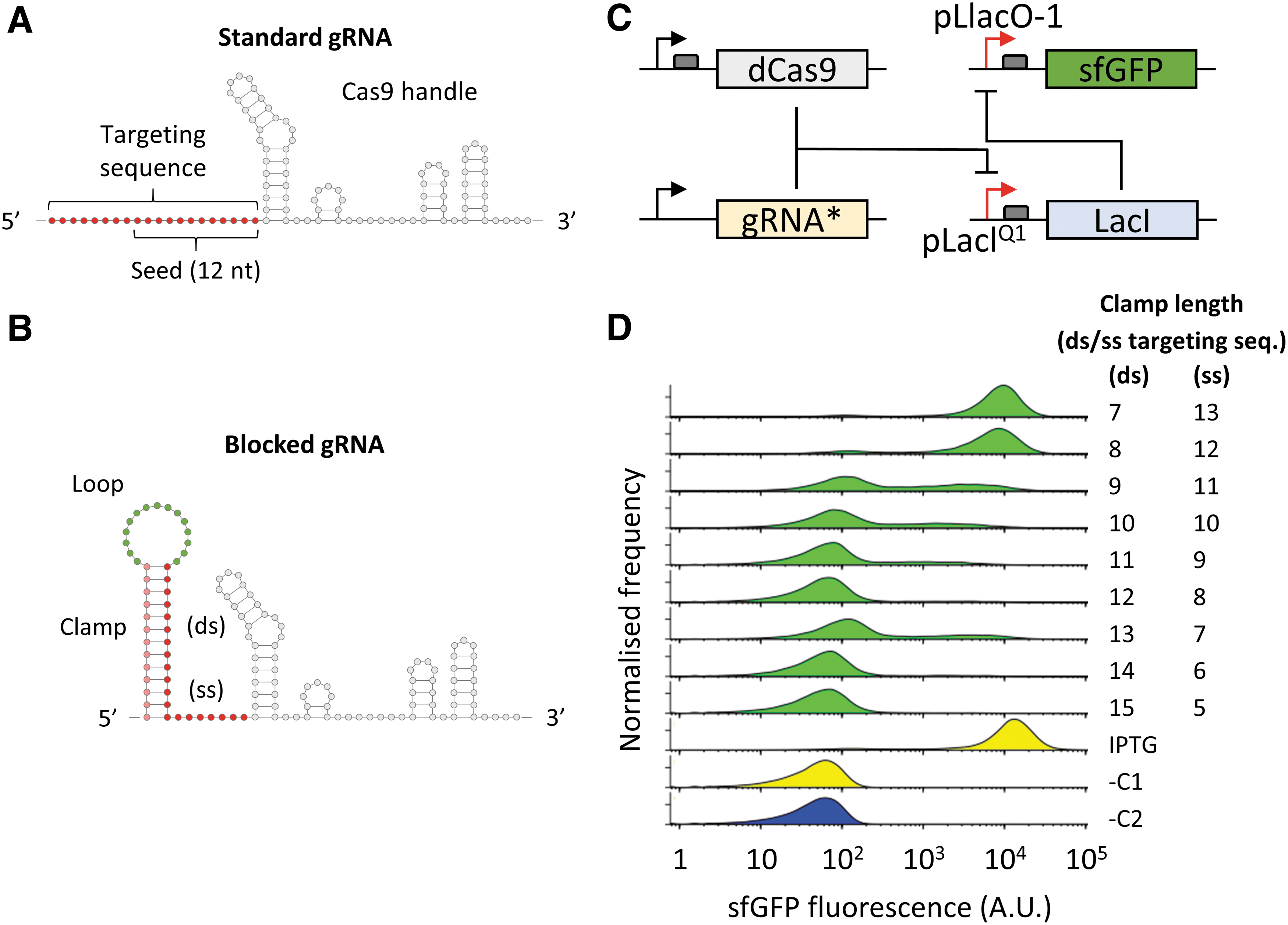
Both the protein and the RNA components of CRISPR-Cas systems have been extensively re-engineered. Specific mutations of the respective nuclease sites generate nuclease-deactivated proteins, such as the most popular dCas9 and dCpf1 (dCas12a), which can be used as RNA-guided DNA-binding repressors for genetic inactivation, also referred as CRISPR interference (CRISPRi). A multitude of other Cas protein variants and fusions have been generated and used for programmable repression or activation of translation as well as for genetic detection.6–9 CRISPR-gRNAs have also been engineered by adding riboswitches 10 or aptazymes 11 to create small molecule-responsive gRNAs, or fused to MS2 or PP7 binding regions, which can be targeted by MS2/PP7 protein fusions.12–15 They can also be incorporated into computational RNA circuits regulated by antisense RNA inhibitors 16 or used for nucleic acid detection.17–19
RNA engineering is greatly facilitated by the possibility of exploiting secondary structure prediction software 20 to create RNA switches that are exclusively activated if in complex with another RNA. This has notably been used for the computational design of riboregulators.21,22 We have previously shown the use of RNA circuitry to engineer logic operations both in vitro and in vivo, either with fully artificial parts or interfacing with natural RNAs. 23 On the other hand, the CRISPR system combines the flexibility of the RNA component, for programmed targeting of virtually any genomic sequence, with various genetic functions allowed by the multitude of CRISPR effectors currently available. This combination offers a virtually infinite number of options for genetic editing and RNA circuit designs.
The rational design of structured sequences in the 5′ region of mRNAs, able to change conformation upon binding of a ligand, has allowed the regulation of gene expression to be engineered entirely within the 5′UTR, without requiring additional protein co-factors. Ligands based on sRNA molecules have been used to design riboregulators and anti-terminators to modulate gene expression.19,21,24–27 We previously proposed a new molecular architecture to engineer RNA-interacting guide RNAs (igRNAs) that change their conformation to the active state exclusively upon hybridization with single-stranded trigger RNAs (trRNAs). 19 Compared to other similar approaches (Supplementary Table S1),28–33 in this work, we aimed to generate flexible igRNA architectures that can be designed with relative ease and virtually no targeting or sensing constraints to activate CRISPR editing upon sequence-specific interaction with synthetic or natural RNA molecules, including full-length and actively transcribed and translating mRNAs in living cells. The absence of interaction between the sensing segment (complementary to the trRNA) and the DNA-targeting sequence of the igRNA allows complete allosteric regulation of CRISPR activity. This means that any trRNA could be used to activate an igRNA targeting a specific genomic sequence and, vice versa, the same trRNA could be used to activate multiple igRNAs targeting different sequences.
We tested our novel RNA-responsive igRNA designs, in both genetic activation and repression settings, upon sensing of sRNA fragments or entire mRNA transcripts with high structural complexity such as those encoding for the fluorescent protein mKate2 and the human immunodeficiency virus (HIV) viral infectivity factor (VIF). We also show proof of principle of adaptability and scalability of igRNA design and testing using fully automatable in vitro cell-free methods for rapid prototyping.
Methods
Plasmid design
For the Escherichia coli assay, the pLlacO-1 promoter 34 was combined with sfGFP 35 within a pSC101-E93K plasmid 36 containing a kanamycin resistance marker. This was further modified using a BioBrick 37 pJ23119-igRNA-HDV ribosome 38 gBlock to create one of the parental plasmids used in this study. The second parental plasmid (Addgene, 46569), 39 containing the dCas9 coding sequence (p15A, chloramphenicol resistance marker). All plasmids used in this study were generated from these two plasmids. For the cell-free transcription-translation (TXTL) assay, the P70a-deGFP plasmid 40 was modified to generate the reporter constructs expressing the deGFP fluorescent protein with the addition of the same CRISPR target sequence used in the E. coli assay at the N-terminus of the deGFP coding sequence. The TX1 reporter carries the target sequence within the deGFP template strand, while the TX4 carries the target in the same position but reverted (non-template strand). The constructs expressing igRNAs, trRNA, or targeting and non-targeting gRNA controls (AJ1–AJ8) were constructed by Golden Gate cloning of each fragment into the receiving vector PAJ486 containing two copies of the J23119 promoter for the co-expression of igRNA and corresponding trRNA (AJ1 and AJ5), targeting gRNA (AJ2 and AJ6) as positive control, non-targeting gRNA (AJ4 and AJ8), and igRNA with the non-corresponding trRNA as negative controls. A detailed description of plasmids, primers, and methods used for their generation is provided in the Supplementary Material (Supplementary Methods and Supplementary Tables S2 and S3).
E. coli assay
E. coli MG1655Z1 cells 41 were grown overnight to early stationary phase at 37°C with shaking in M9 minimal media 21 with the addition of appropriate antibiotics (anhydrotetracycline at a concentration of 100 ng/mL). Cells were then diluted 1:200 in 2 mL of M9 minimal media with the addition of antibiotics and anhydrotetracycline and grown at 37°C with shaking for 2 h before being added to a black-wall clear-bottom 96-well plate. The plates were incubated in a Tecan F500 fluorescent plate reader at 37°C with shaking. A time series of optical density (600 nm) and fluorescence (480/20 nm excitation and 530/25 nm emission for sfGFP, 580/20 nm excitation 635/35 nm emission for mKate2 42 ) readings were taken at 15 min intervals. An average of measurements taken from three biological repeats, each with three technical repeats, was calculated and reported together with the relative standard deviations. All measurements were blanked against M9 media (containing antibiotics and supplements) and against MG1655Z1 cells containing empty plasmids with the appropriate antibiotic markers without igRNA/trRNA/dCas9/sfGFP components to acquire basal fluorescence levels.
Flow cytometry measurements were performed using a BD LSRFortessa (BD Biosciences). Single colonies were grown overnight in LB supplemented with 17.5 μg/mL kanamycin, 50 μg/mL spectinomycin, and 17.5 μg/mL chloramphenicol. The next day, cultures were diluted on a 96-well plate (5 μL in 195 μL), grown (4 h, 37°C, 200 × g), and then diluted for a second time following the same procedure. Once a cell density corresponding to OD600 = 0.3 had been reached, 10 μL of the culture was diluted in 190 μL phosphate-buffered saline (with 2 mM kanamycin to block further protein synthesis). Samples were kept on ice and measured the same day. Flow cytometer voltage settings were: FSC, 632; SSC, 237; B485-530/30, 509. For all the samples tested, FCS data were gated to remove cell debris, and fluorescence histograms were plotted with the Flow Cytometry GUI for Matlab.
Quantitative polymerase chain reaction analysis
Total RNA was extracted from 1.5 mL bacterial cells (OD600nm = 0.6) using a standard Trizol (Thermo Fisher Scientific) protocol. Extracted RNA was precipitated with isopropanol, washed in 70% ethanol, and stored in DEPC-water. The RNA was then treated with DNase I for 1 h at 37°C, re-extracted with Trizol and resuspended in DEPC-water. RNA was tested for DNA contamination using the primers used for quantitative polymerase chain reaction (qPCR). No PCR products were found, indicating the absence of DNA contamination. cDNA (20 μL final volume) was produced from 1 μg of RNA (ReadyScript cDNA synthesis mix; Sigma–Aldrich) and diluted 1:10 with DEPC-water. Diluted cDNA (0.5 μL) was used to confirm cDNA integrity. A qPCR master mix was created according to the manufacturer's specifications (KiCqStart SYBR Green qPCR ReadyMix Low ROX; Sigma–Aldrich) by adding the premixed primer pairs and cDNA (or samples containing no cDNA, and non-retrotranscribed RNA as controls). The qPCR reactions were performed on a Stratagene MX3005p qPCR machine with the following protocol: 1 min at 94°C followed by 40 cycles (15 s at 94°C, 10 s at 57°C, and 20 s at 72°C), melt analysis, and cooling to 30°C. Results were normalized to the expression of the chloramphenicol resistance gene present in the plasmid.
Gel-shift assay
The 3A igRNA RNA fragment was in vitro transcribed (TranscriptAid T7 High-Yield; Thermo Fisher Scientific) from a PCR product containing the T7 promoter upstream of the 3A igRNA. The resulting RNA was extracted with Trizol, precipitated with isopropanol and ethanol, washed and re-suspended in DEPC-water. This RNA (1 μg) was mixed with 1 μg of synthesized 3A or 2A trRNA oligonucleotides (Integrated DNA Technologies), incubated at room temperature for 10 min, and run on a 12% polyacrylamide gel for 2.5 h. The RNA was visualized on a UV transilluminator after staining with ethidium bromide for 20 min.
Cell-free automated assay
Cell-free reactions were performed using 5 nM of each construct in a total volume of 4 μL per reaction. Each reaction contains a combination of three constructs: (1) TX1 or TX4 deGFP reporter; (2) Cas9 or dCas9 plasmid; and (3) plasmid carrying both igRNA (replaced with targeting or non-targeting gRNAs as positive or negative control) and trRNA (2A or 3A). After purification with magnetic beads (AMPure XP; Beckman Coulter), plasmids at standard concentrations were added to 3 μL myTXTL master mix (507024; Arbor Biosciences) and distilled water to a final volume of 4 μL. Each component was transferred from 384-Echo microplates (PPL-0200; Labcyte) into Greiner black small-volume low-base 384-well plates (788876; Greiner) using the acoustic liquid handler (Echo 525; Labcyte) as per the manufacturer's instructions. The Synergy Mx microplate reader (BioTek) was used to analyze deGFP fluorescence (485/20 nm excitation and 528/20 nm emission) every 3 min for 234 acquisitions (∼12 h) at 29°C. A total of 12 replicates were performed for each combination of plasmids.
Results and discussion
5′ added cis-regulatory sequence dictates mechanism of CRISPR-gRNA regulation
With the aim of blocking gRNA annealing to the complementary DNA target, we initially introduced a stem-loop structure to the 5′ end of the 20 nt targeting region of a LacI promoter-targeting gRNA. The structure includes a predesigned 15 nt variable sequence loop 22 preceded by a 7–15 nt long “clamp” sequence complementary to the seed region of the gRNA (Fig. 1B). To test the repression of gRNA binding activity in E. coli cells, we used a reporter system where the gRNA targets the LacIQ1 promoter controlling the expression of the LacI repressor. This was coupled with a sfGFP fluorescent reporter under the control of the IPTG-inducible pLlacO-1 promoter and the dCas9 protein under the endogenous S. pyogenes promoter. In the resulting sensor circuit, the fluorescence signal, normally repressed, is reactivated as a result of CRISPR activity (Fig. 1C).
Upon assay in E. coli cells, we found that the addition of a 7–8 nt clamp, predicted to leave the 12 nt seed region of the gRNA fully exposed, resulted in strong sfGFP signal comparable to the positive control (cells induced with IPTG), indicating successful inhibition of the LacI repressor by the gRNA–dCas9 complex. These results indicate that the 5′ end of the gRNA can accommodate additional nucleotides and secondary structures without affecting its functional activity. On the other hand, clamp lengths hybridizing with ≥9 nt of the targeting sequence resulted in a marked reduction of fluorescence with an increasing proportion of non-fluorescent cells up to levels comparable to the control measurements. These results indicate that a folded RNA secondary structure immediately upstream of the targeting region can generate an active or inactive gRNA, depending on the length of the hybridizing sequence (Fig. 1D).
CRISPR-igRNAs allow allosteric sRNA-responsive modulation of genetic activation or repression in E. coli
In order to transform the 5′ blocked gRNA into a robust allosteric RNA sensor, we added an additional “toehold” structure to the 5′ end, which includes: two variable regions (VR) complementary to the 5′ and 3′ end of the trRNA; the 15 nt toehold sequence (VR1); and the 15 nt loop (VR2), flanking a segment (clamp) that is complementary to 13 nt of the 20 nt targeting region. The resulting igRNA incorporates two discrete parts that are fully independent of each other: the first encompassing two variable regions complementary to the trRNA sequence, including toehold (V1) and loop (V2); and the second constituted by the clamp sequence blocking the 20 nt targeting region in the igRNA inactive state (Fig. 2A). We used the publicly available NUPACK webserver 43 to analyze 30–40 nt segments of the mKate2 sequence and to select two 39 nt long trRNA sequences (identified as 2A and 3A) based on free and co-folding energy with their cognate igRNA molecules as well as reduced self-complementarity at 37°C. Simultaneously, the corresponding igRNA sensors (including toehold, clamp, and loop region) were analyzed to verify minimal complementarity with the downstream secondary structure of the gRNA. The 3A igRNA was initially tested in E. coli using the same LacI-repressed sfGFP reporter used for the clamp testing, with the conditional addition of the trRNA to activate fluorescence expression as a result of CRISPR-igRNA-guided inhibition of the LacI repressor (Fig. 2B). The 3A igRNA showed minimal levels of activity in the absence of the trRNA comparable to the negative controls where the igRNA or the dCas9 constructs were not added. Interestingly, the addition of the corresponding 3A trRNA resulted in high levels of sfGFP fluorescence, corresponding to a 26-fold increase compared to the control experiment where the trRNA was not added. To test the specificity of igRNA activation in blocking the LacI repressor, we also tested the 3A igRNA in the presence of the non-complementary 2A trRNA, which showed only a small increase of fluorescence (1.6-fold) compared to the control without trigger (Fig. 2C). Specificity of hybridization between sensor and trigger sequences was also confirmed by in vitro gel-shift assay where the 3A igRNA showed reduced mobility only in the presence of the cognate 3A trRNA (Supplementary Fig. S1).
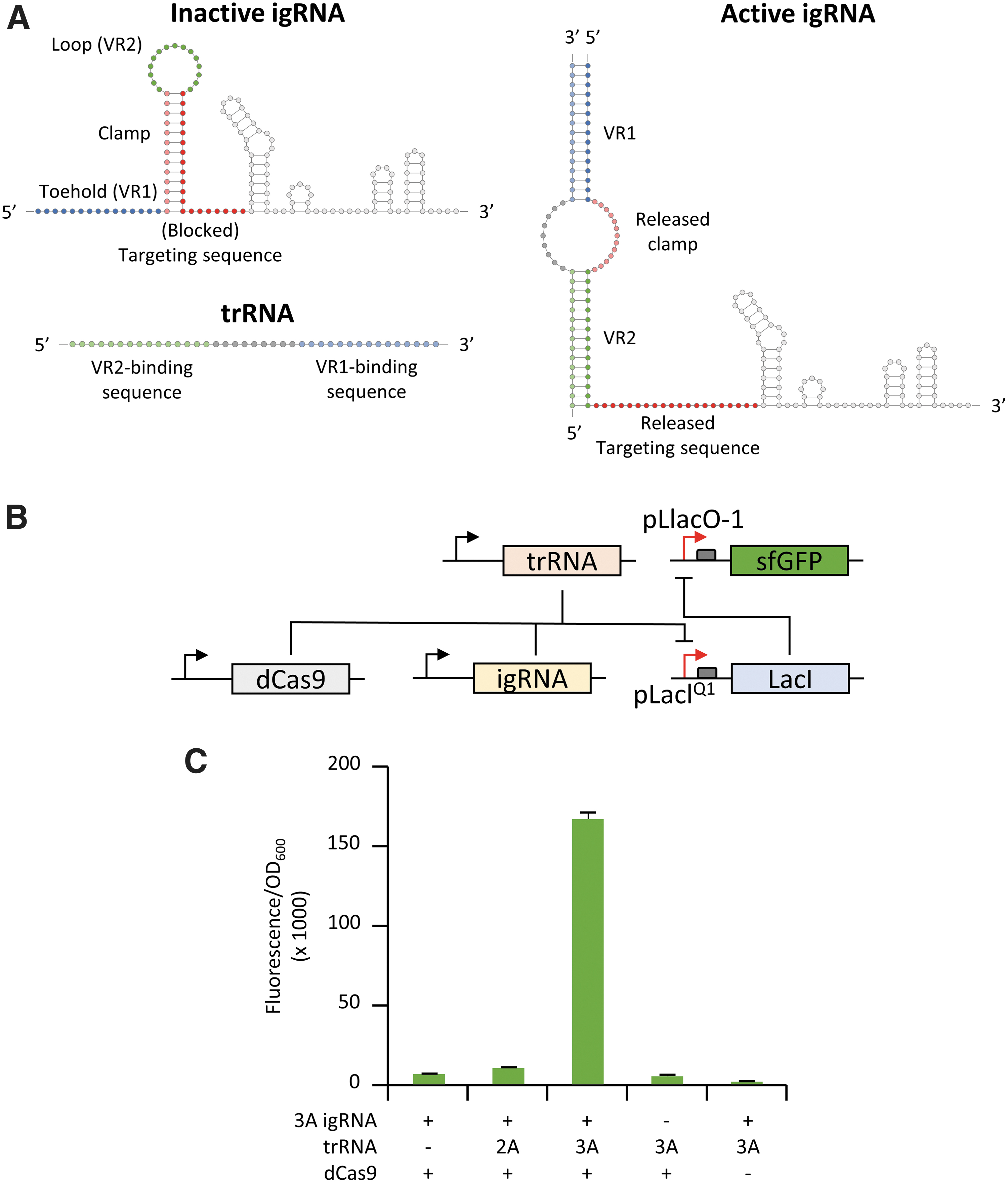
To further explore the adaptability of our igRNA designs, we converted the E. coli-based gene-activating reporter into a genetic repression system. A new hybrid promoter was constructed by fusing a copy of the 20 nt pLacIQ1 targeting region at either side of a computationally designed constitutive promoter devoid of known repressor binding sites (i.e., tetO and lacO) and predicted to promote high levels of expression. 44 This configuration allows the binding of the CRISPR RNP complex to either side of the promoter to improve transcriptional repression when the dCas9 is combined with an actively binding gRNA or trigger-activated igRNA. 44 The hybrid promoter was inserted upstream of a strong ribosomal binding site (RBS) to drive sfGFP expression, and the resulting reporter plasmid (pHyb-sfGFP) was used to replace the LacI repressor and LacIQ1-driven sfGFP constructs (Fig. 3A). E. coli testing of CRISPR-igRNA activity in the new gene-repression architecture showed high levels of sfGFP expression from the hybrid promoter when no trRNA was added, while combining the previously tested 3A igRNA and cognate 3A trRNA generated a strong reduction (∼35-fold) of sfGFP expression (Fig. 3B).

Altogether, these results show that CRISPR-igRNAs can be interchangeably utilized for RNA-dependent genetic repression or activation in live cellular environments.
CRISPR-igRNAs exhibit selective activation by full-length actively transcribed and translating mRNA
After the successful demonstration of sRNA-specific activation of igRNA, we tested whether full-length actively transcribed and translating mRNAs could function as triggers for CRISPR activation in E. coli cells. We initially used the mRNA of the red-fluorescent protein mKate2 to allow visual confirmation of parallel translation of the mRNA trigger via production of the red fluorescent signal. The secondary structure of the mKate2 mRNA transcript was interrogated by using the NUPACK tool to select single-stranded RNA regions. A 39 nt sequence and the cognate igRNA switch (3AV9) were designed following the same architecture used for the 3A igRNA. E. coli testing using the genetic activation reporter (described in Fig. 2B) showed a ∼2.1-fold increase in sfGFP production upon addition of the mKate2 mRNA compared to the control where the mKate2-expressing construct was not added. In order to test whether interactions between the mRNA trigger and cellular translational machinery may interfere with the hybridization and consequent activation of the igRNA, we generated a second version of the mKate2 trigger construct by deleting the respective RBS. As expected, the addition of the translationally inactive mKate2 mRNA showed only basal levels of mKate2 fluorescence. However, sfGFP fluorescence was maintained at similar levels compared to the translationally active trigger, indicating that active translation did not affect binding and activation of the cognate 3AV9 igRNA (Fig. 4A).
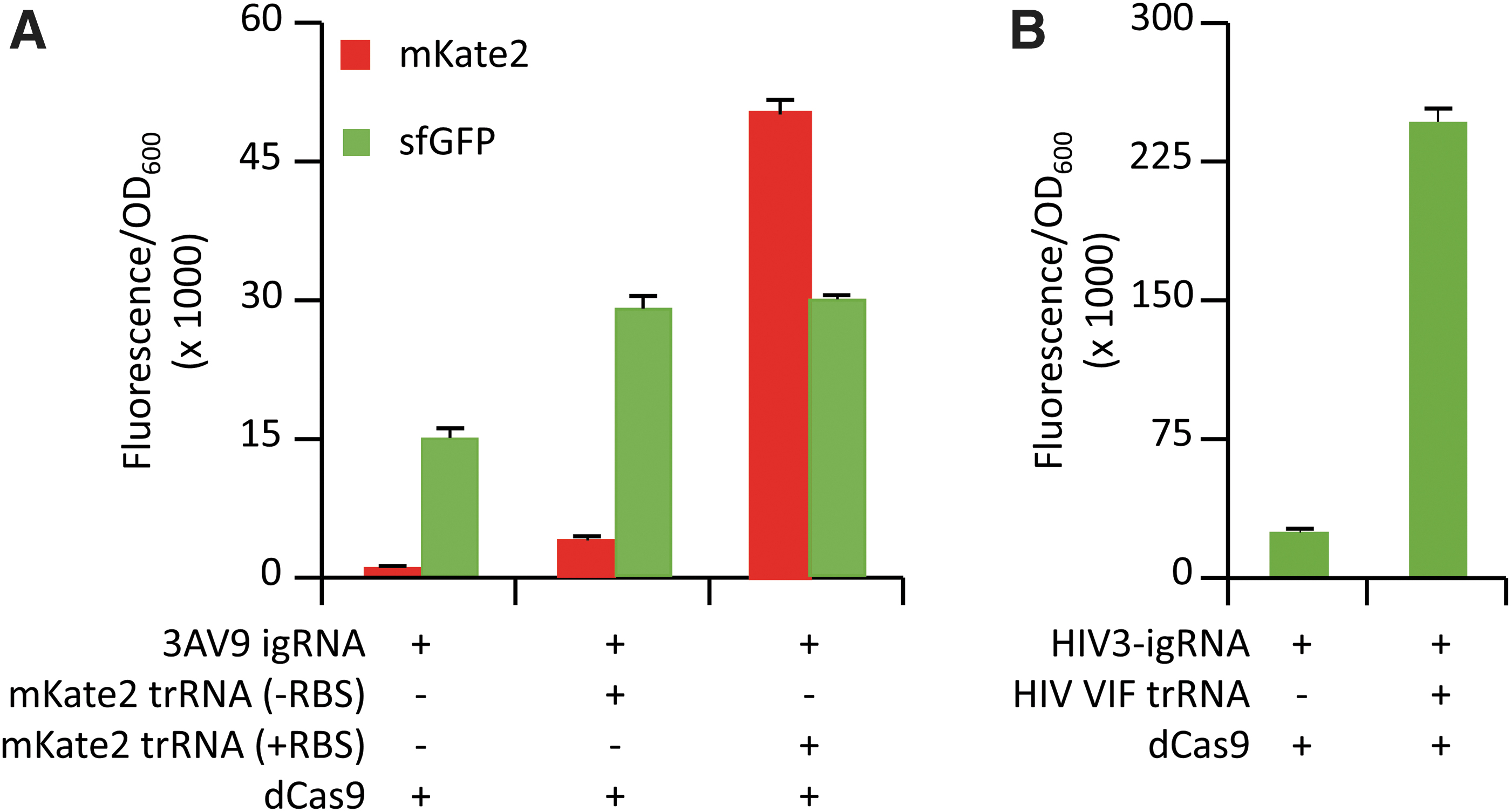
Following on from these results, we decided to test the ability of other medically relevant mRNA transcripts to activate our igRNA designs. For this purpose, we selected the highly conserved VIF mRNA, which is known to be essential for HIV infectivity by playing a critical role in the production of infectious HIV virions.45,46 The computational design of the HIV VIF-responsive igRNA was performed following the same method used for the mKate2 3AV9 switch to select a 34 nt region of the corresponding mRNA (named HIV3-igRNA). The subsequent testing for genetic repression in E. coli reporter showed a consistent increase in sfGFP production (∼10-fold) in the presence of the HIV VIF mRNA compared to the negative control where the viral mRNA trigger was not added, indicating the successful activation of CRISPR-igRNA activity. The concomitant transcription of the HIV VIF messenger transcript in the tested samples was confirmed by qPCR analysis, supporting the ability of HIV3-igRNA to switch to its active form upon interaction with the transcribed viral mRNA (Supplementary Fig. S2).
Scalable in vitro screening of CRISPR-igRNA repression and cleavage activity
After showing that igRNAs can be used to direct dCas9 activity in E. coli cells upon the addition of either short sRNA or long mRNA triggers, we tested the activity of both 3A and 2A igRNAs with either the nuclease-deactivated dCas9 or the nuclease-active Cas9 through an automatable and easily scalable platform for in vitro assay. We generated constructs expressing the igRNA (or standard gRNA as control) and trRNA as well as a customizable reporter construct where the target site for CRISPR-directed cleavage (e.g., using Cas9) or repression (e.g., using dCas9) can be easily interchanged. For this assay, we generated two variants of the fluorescent reporter, including the same target site used in the in vivo testing positioned in both orientations, in order to test CRISPR-igRNA targeting of both template and non-template DNA strand (Fig. 5A). We used an acoustic liquid handler (Echo 525; Labcyte) to set up reactions rapidly in volumes as small as 4 μL on 384-well plates, combining Cas9, igRNA, and trRNA constructs with a commercially available E. coli-based cell-free transcription-translation master mix (myTXTL; Arbor Biosciences). A significant reduction of signal, compared to the negative control, was revealed when both 3A igRNA and 3A trRNA were added to the reaction with either dCas9 (3.7-fold reduction) or Cas9 (3.4-fold reduction) to target the non-template strand of the deGFP reporter. Similar results were also obtained with the 2A igRNA and cognate trigger, giving a 3.6-fold reduction with dCas9 and 4.2 when used with active Cas9. On the other hand, in vitro testing of both 3A and 2A igRNAs showed leaky activity when combined with the non-cognate triggers, ranging from a 1.6-fold reduction when the 3A igRNA was combined with the 2A trRNA to 2.8 when the 2A igRNA was combined with the 3A trRNA for non-template targeting with active Cas9. However, we are aware that the 2A and 3A igRNA tested here were designed based on folding prediction at 37°C, while the in vitro cell-free assay was performed at the recommended temperature of 29°C. 47 As expected, the standard gRNA (without the additional sensor sequence included to the 5′ to generate igRNAs), used as a positive control, gave a strong reduction of fluorescence signal when used to target either non-template or template strand with Cas9, as well as when targeting the non-template strand with dCas9. On the contrary, targeting of the template strand with dCas9 showed only moderate levels of repression, even when using the control gRNA (Fig. 5B and Supplementary Fig. S3). These results confirm previous studies showing that non-template strand binding of the dCas9–gRNA RNP complex is usually required to prevent inhibition or removal from the progressing RNA polymerase and efficiently block elongation of transcription via CRISPRi.7,39 Moreover, the approach proposed here offers the opportunity to scale the throughput of igRNA and cognate trRNA screenings by using a fully automatable platform (Fig. 5C).

Conclusion
The work presented here demonstrates that CRISPR-gRNAs can be converted into cis-repressed igRNAs to transform standard CRISPR-Cas enzymes into freely programmable genetic editors that are precisely activated by specific RNA sequences. The added switch sequence blocks the igRNA ability to bind the targeted DNA sequence, maintaining the Cas9–igRNA complex functionally inactive in the absence of the trRNA. We used igRNAs to switch CRISPR activity, in both in vitro and living E. coli settings, to accomplish genetic repression or activation when in combination with either nuclease active or inactive Cas9 proteins. We show that the interaction between the cis-repressed riboswitch sequence and the cognate trigger can be computationally predicted to engineer igRNAs, showing up to 35-fold differential activation of reporter genes in living cells while maintaining very low baseline activity. The igRNA mode of action relies primarily on RNA folding and RNA–RNA interactions added to the gRNA-5′ end, thereby simplifying design steps and adaptability to the many CRISPR-Cas variants currently identified or engineered, significantly expanding the range of future applications. For this reason, we believe that our CRISPR-igRNA methodology is generalizable to any prokaryotic and eukaryotic host where the CRISPR system is known to work. Importantly, our igRNA architecture offers virtually complete decoupling in the design of the DNA-targeting sequence and the trigger-complementary riboswitch sensor of the igRNA. This distinctive feature opens to the possibility of targeting any desirable gene upon sequence-specific activation by any desirable RNA molecule, pending adequate prototyping and testing. Additional testing will be required to characterize trRNA/igRNA dose–response dynamics of CRISPR-igRNA according to specific applications and corresponding parameters (e.g., temperature, salt concentration, and coexisting RNAs). Nonetheless, our results reveal that igRNAs maintain a high degree of sensitivity in response to small RNA fragments, as well as full-length and actively transcribed and translating mRNAs; for example, the one encoding for the viral infectivity factor of HIV, showing up to 10-fold activation in our in vivo E. coli–based reporter. In addition, we described an innovative platform, combining fully customizable and automatable in vitro reporter assays, for high-throughput screening and optimization of CRISPR-igRNA components supporting future development of our technology. We suggest that the methodology described here could be used for the de novo design and testing of various in vitro or cellular sensors for a multitude of applications, including the development of new diagnostic and therapeutic tools.
Footnotes
Acknowledgments
The authors would like to thank the Warwick WISB Centre for the use of the BD LSRFortessa cell analyzer; the Crisanti Lab and the London Biofoundry at Imperial College London for support with the cell-free assay and use of the Echo 525 acoustic liquid handler. We would also like to thank M.B. Elowitz for the MG1655Z1 strain.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the 7th Framework Programme (grant numbers 610730 [EVOPROG], 613745 [PROMYS]); the Horizon 2020 Marie Sklodowska-Curie (grant number 642738 [MetaRNA]); the Engineering and Physical Sciences Research Council (EPSRC) and the Biotechnology and Biological Sciences Research Council (BBSRC; grant numbers BB/P020615/1 [EVO-ENGINE] and BB/M017982/1 [WISB Centre]); and the School of Life Sciences (University of Warwick; start-up allocation) to A.J. Funding for open access charge: EPSRC/BBSRC (BB/M017982/1) to A.J. W.R. was supported by a DGA-Dstl fellowship.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.