
Abstract
CRISPR- Cas9 has revolutionized genetic engineering. However, the inability to control double-strand break (DSB) repair has severely limited both therapeutic and academic applications. Many attempts have been made to control DSB repair choice. However, particularly in the case of larger edits, none have been able to bypass the rate-limiting step of homologous recombination (HR): long-range 5′ end resection. Here, we describe a novel set of Cas9 fusions, Cas9-HRs, designed to bypass the rate-limiting step of HR repair by simultaneously coupling initial and long-range end resection. Here, we demonstrate that Cas9-HRs can increase the rate of homology directed repair (HDR) by 2- to 2.5-fold and decrease p53 mediated cellular toxicity by two- to fourfold compared to Cas9 and are functional in multiple mammalian cell lines with minimal apparent editing site bias. These properties should make Cas9-HRs an attractive option for applications demanding increased HDR rates for long inserts and/or reduced p53 pathway activation.
Introduction
CRIPSR-Cas9 technology has revolutionized genetic engineering by allowing quick and facile targeting of virtually any accessible spot in the genome via RNA-based guides.1–3 CRIPSR-Cas9 generally has two broad uses in genome editing: mutation of targeted sites via imprecise non-homologous end joining (NHEJ)-mediated repair and introducing precise edits in the genome ranging from 1 to 1,000s of base pairs (bp) using an external template sequence via homology directed repair (HDR). 4 While HDR methods generally allow for more flexible editing, NHEJ repair is highly preferred in higher eukaryotes where HDR repair rates can be as much as two orders of magnitude less than NHEJ repair. 5 Low HDR/INDEL ratios have so far generally limited therapeutic applications of CRISPR-Cas9 to only those which are amenable to NHEJ-based repair and makes the introduction of targeted mutations, insertions, or deletions difficult, expensive, and time consuming.6,7 In addition, NHEJ-based repair can activate the p53 pathway, prolonged activation of which can lead to cellular apoptosis, not only reducing yields of edited cells but also potentially selecting for cancerous mutations. 8
There are many different approaches that have been taken to attempt to improve HDR rates. However, most fall into four broad classes: small molecule and genetically encoded inhibitors or activators of NHEJ and HDR, respectively, cell-cycle synchronization, optimization of culture conditions, and engineering of the nuclease(s) themselves.9,10 While small molecule and genetically encoded inhibitors can be effective in vitro, these techniques must be optimized for each cell type and can have undesirable side effects that significantly limit their usefulness. As such, significant effort has been put into engineering Cas9 (and other nucleases) to provide more flexible alternatives, and while there are still some issues with these tools that need to be resolved, there is no doubt that they have dramatically impacted the landscape of genetic engineering and point to the significant benefits that engineered Cas9 fusions can bring.11–13
Unfortunately, Cas9 fusions designed to increase the HDR rate for longer inserts (>0.3 kb) have not been as successful, though recently fusion of various factors involved in double-strand break (DSB) repair choice (CtIP, Mre11, and a truncated piece of p53 named DN1s) to Cas9 have been shown to increase the HDR:INDEL repair ratio for longer inserts from ∼1.5- to 2.5-fold.14–16 However, each of these tools still has significant issues. Cas9-CtIP has been shown to have both editing site and cell type bias, and since both CtIP and Mre11 undergo extensive post-translational regulation and have a myriad of protein–protein interactions, it is likely that these issues are related to endogenous cellular components themselves.17–19 Additionally, none of these fusions have directly been shown to reduce cellular toxicity. In fact, the most effective in terms of HDR:INDEL ratio increase, Cas9-DN1s, actually increase toxicity by roughly 5–10%. 16 Equally important, their mechanisms of action fundamentally limit their effectiveness to endogenous rates of HDR repair, as none of these fusions act on what is thought to be the rate-limiting step: long range 5′ → 3′ end resection. 18
Eukaryotic DSB repair takes place via two main pathways: canonical and non-canonical. The first relies on binding of MRN/CtIP complex slightly downstream of the DSB, which then resect back via 3′ → 5′ exonuclease activity to create short (<20 bp) single-strand (ss) DNA 3′ ends. After additional steps, the overhanging ssDNA 3′ ends are eventually further resected via long-range 5′ → 3′ exonucleases Exo1 or Dna2, which then commits the DSB to be repaired via HR. 19 The non-canonical pathway simply bypasses the initial 3′ → 5′ resection, with either Exo1 or Dna2 directly initiating and resecting the DSB 5′ ends to create long 3′ ssDNA ends, thus committing the DSB to HR repair. It is thought that the canonical repair pathway prevails due to DBS ends being “blocked” by other bound proteins. 20 However, fusion of either Exo1 or Dna2 to Cas9 should allow them preferential access to the DSB, in theory greatly increasing the chance of committing the DSBs to HR via the non-canonical repair pathway. While both human Exo1 and Dna2 were considered as initial fusion partners, ultimately hExo1 was chosen to test as the initial fusion partner due to the availability of significantly more biochemical and structural data compared to Dna2.17,21
Methods
Genomic DNA extraction, amplification, and sequencing
DNA was extracted from cells using the DNeasy mini kit (Zymo Research) per the manufacturer's instructions. DNA was then amplified with either standard Taq (Bioneer), Fusion (NEB), Q5 (NEB), or PrimeSTAR GXL (Takara) using standard polymerase chain reaction (PCR) protocols. Genome integrated H2B-RT-3′ was amplified with standard Taq and primers hH2B-3′-F and -R at 56°C for 35 cycles; H2B-RT-5′ was amplified using PrimeSTAR GXL and primers hH2B-5′-F and -R at 65°C for 35 cycles. Genome integrated Puro RT-3′ was amplified with Phusion polymerase and Puro-Int-3′-F and -R at 58°C for 35 cycles; Puro RT-5′ was amplified with Phusion polymerase and Puro-Int-5′-F and -R at 56°C for 35 cycles. Genome integrated Renilla Luciferase cassette (RLucRT) was amplified using nested PCR with Primestar GXL. The first round was performed using Rluc_G-5′-F2 and Rluc_G-5′-R2 for the 5′ junction and RLuc_G-3′-F1 and Rluc_G-3′-R for the 3′ junction at 64°C for 30 cycles. Reactions were then diluted 1:200, and the second round of PCR was performed for 5′ with Rluc_G-5′-F1 and avRT-rLuc-P2-R, and for 3′ with Rluc_G-3′-F3 and Rluc_G-3′-R2 at 62°C for 30 cycles. All 5′ and 3′ integrated amplicons were then gel extracted using the QIAquick Gel Extraction kit (Qiagen), and then sequenced from both ends via sanger sequencing. Sequences were inspected and visualized with ApE and Jalview.
Cell culture and transfection and drug delivery
Unless otherwise noted, adherent cells (A549 or H1299) were seeded on 96-well plates and grown in either 50/50 F-12/Dulbecco's modified Eagle's medium or RPMI-1640, respectively, supplemented with 5% fetal bovine serum (FBS) and grown to roughly 70% confluency. Cells were then transfected with either Cal-Phos, as described in Chen et al., 23 or Lipofectamine 3000 (Thermo Fisher Scientific). Fresh media was exchanged the next day, and cellular viability was quantified on day 2.
K562 cells were grown on 24-well plates with RPMI-1640 supplemented with 5% FBS until roughly 70% confluent. At that time, cells were electroporated using either the mNeon system (Invitrogen) with optimized settings for K562 cells, or Lipofectamine 3000, again following the manufacturers' instructions. Cells were then returned to 24-well plates with fresh media and grown for at least 2 days before being used in downstream analysis.
Pifithrin-α (MilliporeSigma) was diluted to appropriate concentrations such that a 1:100 dilution resulted in the desired final concentration and was applied concurrent with transfection, with equal amounts of dimethyl sulfoxide (DMSO) added as a control.
Cellular toxicity quantification
Resazurin solution (20 μL of 0.15 mg/mL) was added to cells plated on 96-well plates for all cell lines, which were then incubated for ∼4 h at 37°C. Cellular toxicity was quantified using a SpectraFlour Plus plate reader (Tecan) using a 535/595 filter set. Raw data were then normalized to the mean of the untransfected controls and plotted as described.
H2B-mNeon knock-in HDR quantification
K562 cells were transfected with 500 ng Cas9-HRs 4, 5, 6, and 8, or Cas9 targeting hH2B-G4, plus 50 ng hH2B-mNeon RT. After 2 days, cells attached to coverslips were coated with 0.01% poly-
Cas9-HR Puro RT HDR q uantification
K562 cells were transfected with 500 ng Cas9-HR 8 or Cas9 targeting Int-G2 or Int-G3 plus 100 ng Puro RT. After 2 days, 1/10 of cells were taken for DNA extraction, while the rest were treated with 0.5 μg/mL puromycin. Cells were grown for a further 3 days and then viability quantified via a resazurin assay (MilliporeSigma).
Cas9-HR and Cas9 luminescence assays
H1299 were seeded, grown on 96-well plates as for other experiments, and then transfected with 500 ng Cas9-HR 8 or Cas9 (NT) targeting either AAAVS1 G1 or G2 with 50 ng AAVS1 RLucRT. After 2 days, cellular viability was quantified via a resazurin assay. Then, cells were washed with PBS and lysed with 25 μL cell lysis buffer. Luciferase activity was quantified via Renilla Luciferase Assay (Promega) and using a Tecan Infinite M plex plate reader.
Results
Initial design and characterization of Cas9-HRs
A fragment of human Exo1 (1-352), which possess exonuclease activity but lacks post-translational regulation, was N-terminally fused to Cas9 with linkers of different lengths and amino acids, as linker identity and length can have a profound effect on fusion protein stability and activity. 22,23 Two different versions of Cas9 were used—one with two nucleus localizing sequences, (2XNLS)-Cas9, and one with the N-terminal NLS deleted, 1XNLS-Cas9—hypothesizing that the extra NLS could interfere with proper fusion of hExo1 (Fig. 1A), and a directly fused hExo1–Cas9 construct was developed as well. Hereafter, the hExo1(1-352)–Cas9 fusions will be referred to Cas9-HRs 1–9. Plasmid PX330 was chosen as the expression vector, as it allows for fascicle simultaneous expression of Cas9 and gRNA (Fig. 1B, top). 1 After constructs were cloned and sequenced, Cas9-HRs 1–8 were then tested in human lung carcinoma A549 cells, which importantly have retained a functional p53 gene. 24 An intergenic region on chromosome 12 was targeted, with the idea that if Cas9-HRs can shift cells from NHEJ to HR repair, p53 pathway activation and corresponding cell death should be reduced compared to unmodified Cas9.
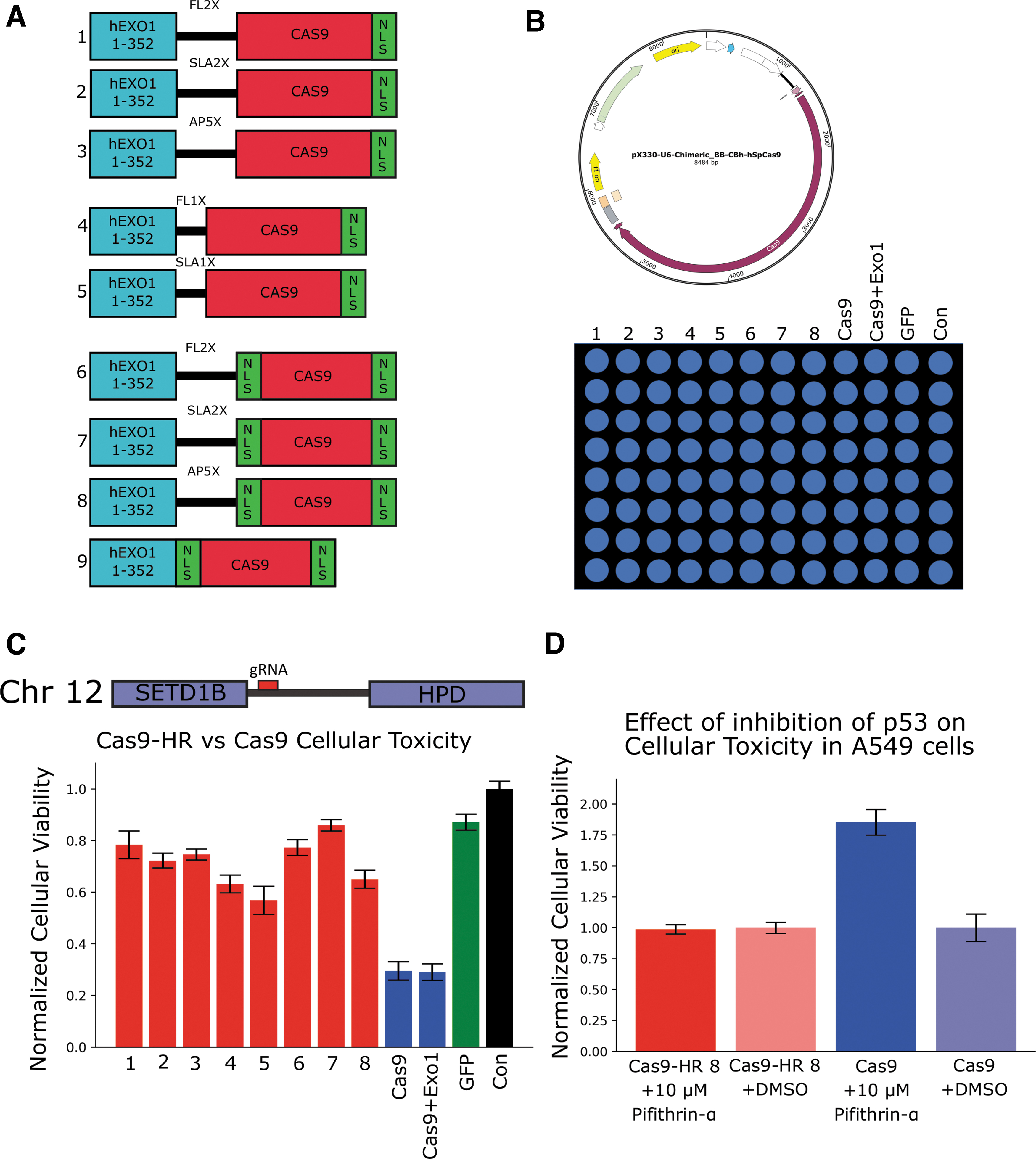
Initial construction and characterization of Cas9-HRs.
Cells were plated on 96-well plates and transfected with Cas9-HRs 1–8, Cas9, and green fluorescent protein (GFP) using a standard calcium phosphate transfection technique and incubated overnight for 16–20 h (Fig. 1B, bottom). Cells were then allowed to recover for one additional day, after which viability was quantified via a resazurin assay and a plate reader (PerkinElmer). All Cas9-HR fusions had greatly increased cellular viability (∼2- to 4-fold) compared to unmodified Cas9, and hExo1 fusion to Cas9 is required for this reduction in cellular toxicity (Fig. 1C). Dramatically, most had similar survival rates when compared to GFP, indicating that Cas9-HRs may significantly reduce NHEJ repair and subsequent p53 pathway activation.
Cas9 cellular toxicity in A549 is in part mediated by the p53 pathway
To probe the role of p53 in Cas9-mediated toxicity, A549 cells were again transfected with Cas9 or Cas9-HR 8 and treated with the cell permeable p53 inhibitor Pifithrin-α (MilliporeSigma). Cells transfected with Cas9 and treated with 10 μM Pifithrin-α had a ∼1.75-fold increase in cellular viability compared to Cas9 treated with DMSO (solvent). Cells transfected with Cas9-HR 8 showed no significant change regardless of treatment (Fig. 1D). Additionally, A549 cells transfected with Cas9 and treated with increasing amounts of Pifithrin-α showed a dose-dependent response in cellular viability (Supplementary Fig. S1A). However, Pifithrin-α treatment could not fully reduce Cas9-mediated toxicity, though this could possibly be explained by incomplete inactivation of the p53 pathway. Regardless, these results indicate that the cause of Cas9 toxicity in A549 cells is at least partly due to activation of the p53 pathway, as seen in other p53+ cell types, and that Cas9-HRs do not seem to activate the p53 pathway to the same extent as Cas9, thereby offering a potential explanation for their increased cellular viability.8,25
Cas9-HRs show full-length expression and correct subcellular localization
To probe Cas9-HR expression and localization, K562 cells on 24-well plates were transfected with Cas9-HRs 4–8 and Cas9 using Lipofectamine 3000, with Cas9-HRs 1–3 omitted due to similar initial reduction in toxicity as Cas9-HRs 6–8 (Fig. 1D). K562 cells were chosen due to their p53–/– status, ideally minimizing the cellular toxicity effects of Cas9 transfection and thus facilitating accurate quantification of expression levels. 26 K562 cells were either lysed with RIPA buffer (Santa Cruz Biotechnology) or fixed with 4% PFA, and then probed for expression levels and sub-cellular localization via an α-Cas9 antibody (Santa Cruz Biotechnology) through both Western blot and frozen-tissue immunohistochemistry (F-IHC). Detectable expression of bands at 200 kD roughly corresponding to full-length Cas9-HR (∼200–205 kD) were seen for all constructs, though with significantly reduced, though still detectable, expression levels for Cas9-HR 4 (Supplementary Fig. S2A, left: top and bottom). Additionally, Western blots of K562 cells transfected with Cas9-HR 8 and Cas9 and untransfected control cells demonstrated the expected ∼40 kD size shift for Cas9-HR 8 compared to Cas9, and this high molecular weight staining was specific, as it was not seen in control cells (Supplementary Fig. S2A, right). Finally, F-IHC experiments further demonstrated detectable expression and proper nuclear localization (white arrows) of Cas9-HRs 4–8 (data not shown for 5–7) comparable to that of Cas9 (Supplementary Fig. S2B). These results demonstrate that Cas9-HRs are properly expressed and localized when expressed in human cells.
Cas9-HRs simultaneously reduce toxicity and increase HDR rate
The next experiments were designed to test both Cas9-HR nuclease activity and effects on HDR editing rate simultaneously, as well as to elucidate further the role of p53 in Cas9-mediated cellular toxicity. First, a repair template designed to tag endogenous H2B with mNeon (referred to hereafter as H2B-mNeon RT) was constructed, including two silent mutations (indicated by the red bar) designed to disrupt Cas9 binding (Fig. 2A). A549 cells were transfected using Lipofectamine 3000 with Cas9-HRs 4, 5, 6, and 8 and Cas9 with H2B-G4 guides. Interestingly, Cas9-HR 8 was the only Cas9-HR to show a significant reduction in cellular toxicity compared to Cas9 (Fig. 2B, left). Next, given that Pifithrin-α treatment in A549 cells likely does not fully inhibit p53 pathway activation, H1299 cells—a lung carcinoma cell line that unlike A549 cells lacks a functional p53 gene—were used as an independent assay to examine further the effect that p53 pathway activation has on Cas9-mediated cellular toxicity. H1299 cells were plated and transfected similarly to A549 cells, and resazurin quantification of cellular viability demonstrated a dramatic reduction in toxicity for Cas9-HRs 4–6 and Cas9 in H1299 cells compared to A549, with Cas9-HR 8 only showing a small reduction (Fig. 2B, right). These results further demonstrate that Cas9-HR toxicity reduction is very likely due to reduced activation of the p53 pathway, indicating Cas9-HRs may be particularly useful in applications where significant p53 activation is undesirable.
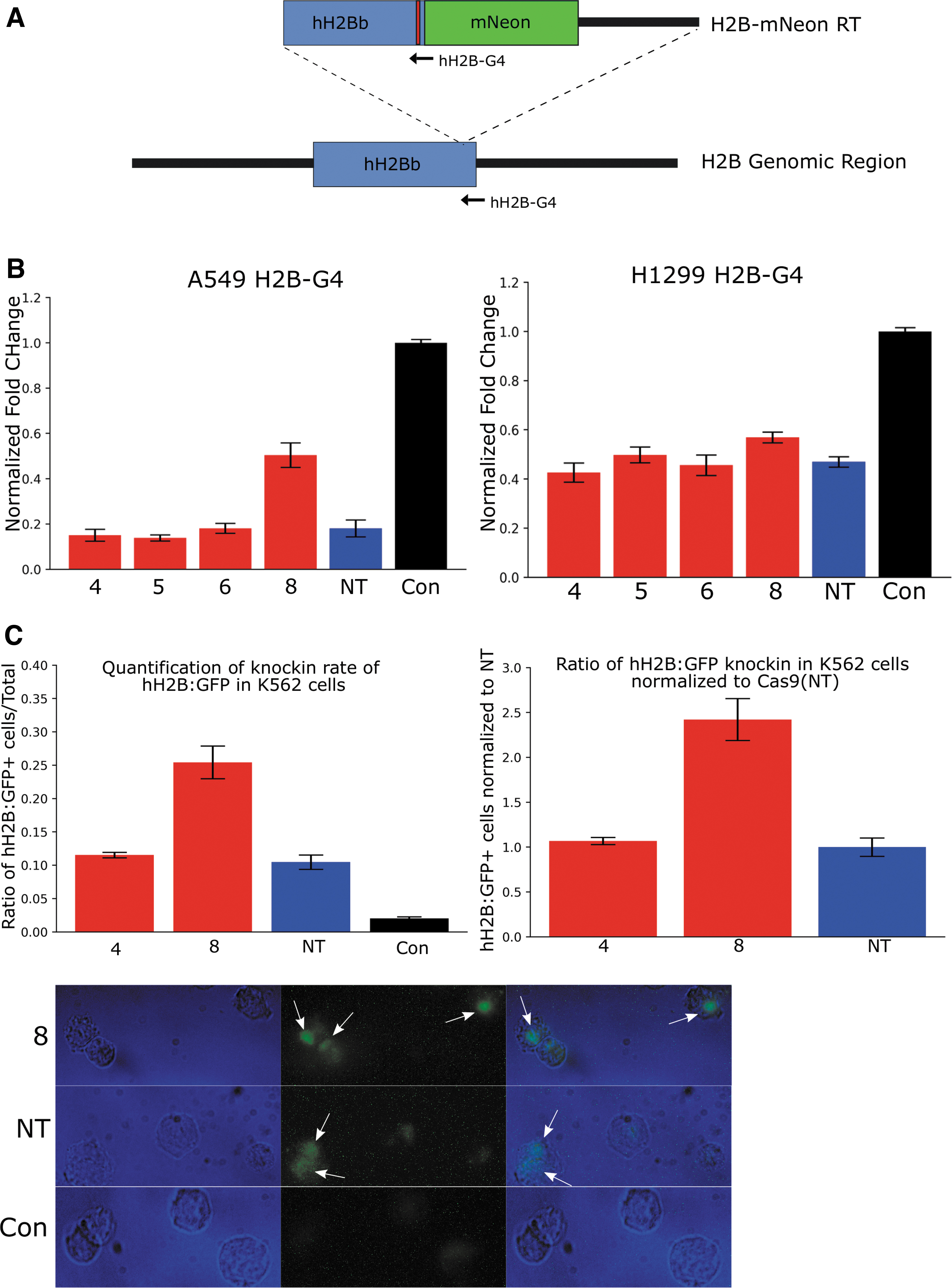
Cas9-HR 8 decreases cellular toxicity and increases rates of homology directed repair (HDR).
Next, to test whether Cas9-HRs can improve HDR repair rates, cells were transfected via electroporation with both H2B-mNeon RT and only Cas9-HRs 4 and 8 and Cas9, since Cas9-HRs 4 and 8 had shown the most promise in reducing toxicity. After 2 days, cells were fixed in 4% PFA, washed in PBS, and then directly imaged, with cells showing nuclear localized fluorescence counted as successful HDR events. As with toxicity reduction in A549 cells, Cas9-HR 8 showed a significant increase in HDR rate compared to either Cas9-HR 4 or Cas9 (Fig. 2C, left and right), with representative fluorescent images of cells classified as HDR positive and negative cells shown in Figure 2C (bottom). These results demonstrate that not only do Cas9-HRs retain nuclease activity, but they may also in fact be able to increase the HDR rate substantially.
To confirm proper tagging of H2B with mNeon, DNA was extracted with the DNeasy mini kit (Zymo Research) from 1/10 of H2B-mNeon RT + Cas9-HR 4 and 8 or Cas9-transfected K562 cells. Regions surrounding the putative knock-in were amplified using 5′ (blue) and 3′ (red) specific primers (Supplementary Fig. S3A), with each primer pair containing one genome specific and one RT specific primer. Successful amplification of both 5′ and 3′ pairs was seen with both Cas9-HR 4 and 8 and Cas9 (NT), but not in untransfected control samples. Additionally, the increase in band strength of Cas9-HR 8 5′ product compared to Cas9-HR 4 or Cas9 further confirms the imaging results that Cas9-HR 8 likely has an increased HDR rate relative to Cas9 (Supplementary Fig. S3B, 3′ appears to be out of quantitative range). The 5′ and 3′ PCR products sequenced, and both Sanger traces and consensus alignments of 4, 8, and NT showed no gross differences in sequence or genomic mutations (Supplementary Fig. S3C). These experiments demonstrate that Cas9-HRs not only can increase the HDR rate but, as expected based on initial design, also appears to maintain functionality across different cell types.
Cas9-HRs Decrease toxicity and increase HDR rates at multiple additional genomic loci
Next, an additional set of experiments were designed to test further the relationship between cellular toxicity and repair pathway choice, as well as to test if Cas9-HRs can increase the rate of HDR for an independent and longer (1.8 kb) insert. First, an HDR template containing a puromycin resistance cassette was created via fusion PCR, with 5′ and 3′ homology arms added respectively as shown in Figure 3A (left). The target integration site is approximately ∼1 kb to the 3′ end of the human H2B gene on chromosome 6—an intergenic region that has no predicted genes or long non-coding RNA. Quantification of toxicity in A549 cells was again determined as in Figure 1C. A549 were transfected via CalPhos with either Cas9-HRs 4 or 8, Cas9 targeting either Int-G2 or Int-3, or Puro RT alone. Significant toxicity was seen with Cas9 targeting both Int G-2 and G-3, while Cas9-HRs 4 and 8 both showed a dramatic reduction in cellular toxicity (Fig. 3A, right). Interestingly, Cas9-HR 4 showed significantly more toxicity targeting G-2 rather than G-3, indicating a potential differential site preference of Cas9-HR 4 compared to Cas9-HR 8.
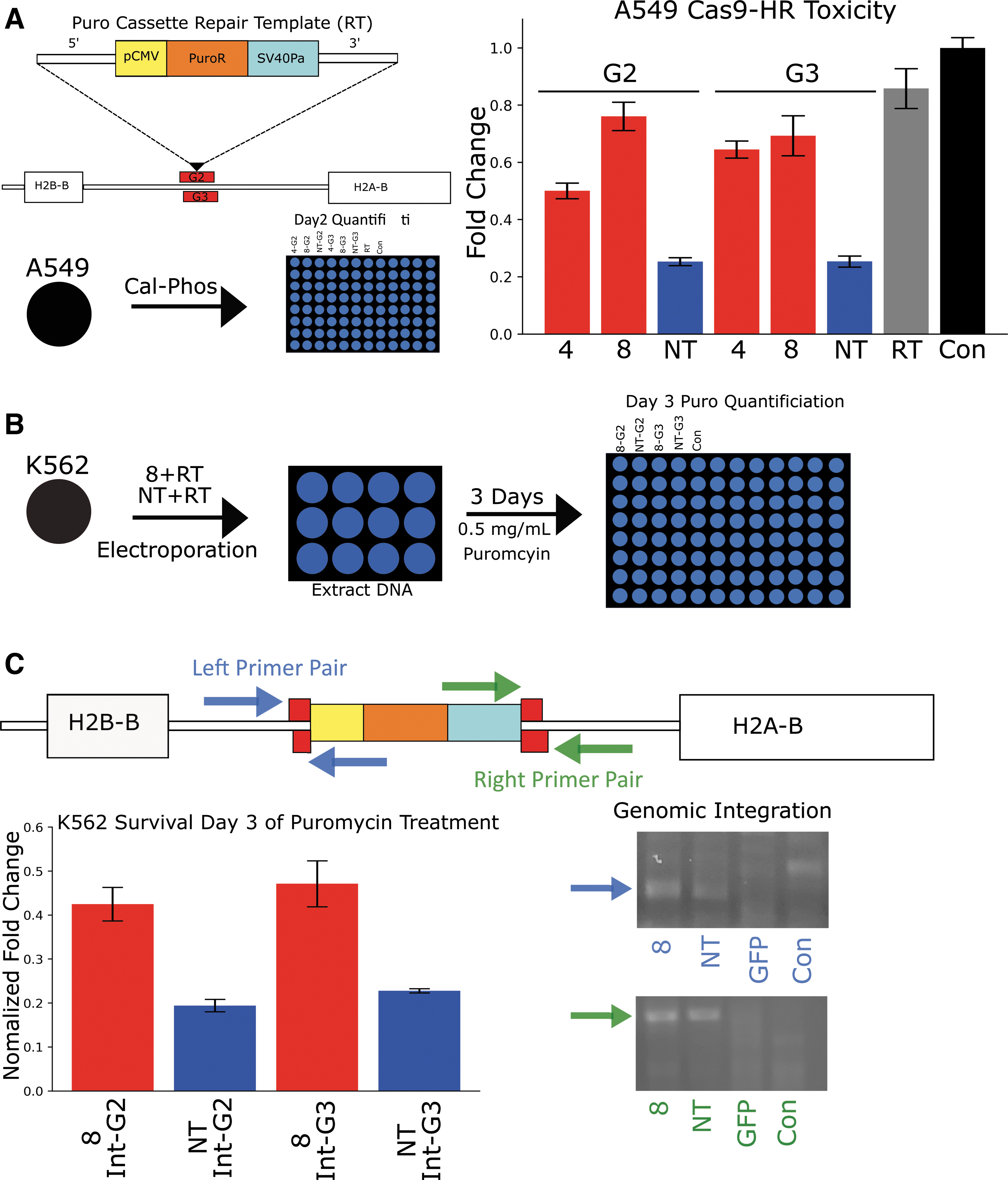
Cas9-HR 8 reduced toxicity and increased HDR rates in an additional independent assay.
Next, in order to assay HDR rates, K562 cells were again used instead of A549 cells, as the lack of a functional p53 gene should help to deconvolute HDR rates from cellular toxicity effects. Additionally, only Cas9-HR 8 was assayed, as it was unlikely Cas9-HR 4 would be superior to Cas9-HR 8 at either locus, given previous toxicity results. K562 cells were grown on 24-well plates, which were then electroporated with Cas9-HRs 8 or Cas9 and 100 ng of amplified repair template (RT), as shown in Figure 3B. After 2 days, DNA was extracted from ∼1/10 of surviving cells and used for analysis of Puro RT genomic integration. The next day, 0.5 mg/mL puromycin was added, and after 3 days, cellular survival was quantified via a resazurin assay, as described previously. As shown in Figure 3C (left), Cas9-HR 8 G-2 and G-3 had around twice as many surviving viable cells compared to unmodified Cas9 (NT). Again, amplification using primers designed specific for the genome (containing no sequence used in the RT) and specific for the RT at both 5′ and 3′ ends demonstrated successful integration of the repair template in both Cas9-HR 8 and Cas9, but not in either GFP or untransfected cells. These results further demonstrate that Cas9-HRs have significantly increased HDR rates relative to Cas9. They also demonstrate cross-functionality across cell types and further link reduction of toxicity to increases in HDR rates.
For an additional comparison of Cas9-HR versus Cas9 HDR rates, both a different integration site and HDR readout assay were used. For these experiments, the well-characterized safe harbor site AAVS1 was used combined with previously established and functional guide RNAs AAVS1 T1 and T2, which are referred to as G1 and G2, respectively, hereafter. 27 A repair template was constructed as shown in Figure 4A, containing 5′ and 3′ AAVS1 homology arms surrounding a constitutively expressed RLucRT. As with the puromycin resistance cassette, PCR-amplified dsDNA was used as opposed to linearized plasmids in order to avoid confounding results when quantifying HDR rates and to avoid extra integration of plasmid-specific sequences in the genome. These experiments also used H1299 cells, as they are both p53–/– and adherent, which should help to minimize experimental error due to differences in Cas9-HR versus Cas9 toxicity and potential handling errors. Finally, since Cas9-HR 8 had shown the best performance with the other various targets tested to date, only Cas9-HR 8 was compared to Cas9 for these experiments.

Cas9-HR 8 has increased HDR activity compared to Cas9 at the AAVS1 safe harbor site.
H1299 cells were transfected with Cas9-HR 8, Cas9 plus RLucRT targeting AAVS1 G1 or G2, RLucRT alone, or control untransfected cells. Next, the viability of transfected cells was quantified via resazurin after 2 days. Then, cells were washed with PBS and lysed, and luminescence was quantified via a plate reader (Fig. 4B). As expected, no gross changes in cellular viability were seen with transfection of either Cas9-HR8 or Cas9 plus RLucRT compared to RLucRT alone (Fig. 4C). Next luminescence was quantified via the Renilla-Glo Luciferase Assay System (Promega) using a 96-well plate reader with luminescence capabilities (Tecan). After data collection, raw luminescence was background subtracted from non-transfected control cells, corrected for cellular viability, and plotted in Figure 4D. While both Cas9-HR 8 and Cas9 targeting AAVS1-G1 showed significantly higher luminescence than AAVS1-G2, Cas9-HR 8 consistently showed significant increases in luminescence (∼2.5- and ∼2-fold, respectively) relative to Cas9 when targeted with either G1 or G2 (Fig. 4D, left, right). As before, integration in the genome was confirmed via junction PCR, with successful amplification for both 5′ and 3′ junctions, with successful amplification seen with all treatments except controls (Fig. 4E and data not shown). Given the extremely sensitive nature of nested PCR, it is not surprising that integrations were detected in the RT samples, even if the amount was too low to be detected via luminescence assay. Sequencing of purified amplicons from each treatment confirmed correct and specific amplifications, with no gross abnormalities seen (data not shown). Combined with previous results, these data strongly indicate that the Cas9-HR series (and Cas9-HR 8 in particular) has significantly higher HDR editing rates than Cas9 and is functional across multiple cell types and genomic loci.
Discussion
The ability to control DSB repair pathway choice has been long sought after, with numerous different tools and techniques developed to influence repair choice. As described here, Cas9-HRs represent the first ever example of a tool designed to act at the rate-limiting step of HR repair.4,5,9 As a first pass for the efficacy of this tool type, HDR assays for Cas9-HRs (particularly Cas9-HR 8) have demonstrated significant increases in HDR rates (∼2- to 2.5-fold) compared to Cas9 at multiple loci across multiple cell and assay types. Interestingly, in addition to increased HDR rates, Cas9-HRs also show significantly reduced activation of the p53 pathway, potentially allowing for extension of high-efficiency HDR methods to more sensitive cell types or even select in vivo applications. It will be particularly interesting to investigate the exact mechanisms behind Cas9-HRs ability to increase HDR rates and reduce cellular toxicity, as this potentially could shed further light on fundamental principles governing eukaryotic DSB repair choice.
Given other experiences with Cas9 fusion proteins, it is likely that further rounds of engineering will be required to improve Cas9-HR efficacy and hopefully boost HDR rates even further. Additionally, since the current size of Cas9-HRs (∼5.5 kb) makes them slightly too large for current adeno-associated virus techniques, developing fusions using other smaller Cas9s or other more compact RNA-guided nucleases in addition to other fusions and mutations designed to expand Cas9-HR functionality and targetability will be a priority.
Conclusion
As demonstrated here, the Cas9-HR platform represents a significant step forward in controlling DSB repair choice and should prove particularly useful for applications demanding increased HDR rates for large inserts and/or reduced p53 pathway activation.
Footnotes
Acknowledgments
The author would like to thank MBCBiolabs and BioCurious for providing the opportunity and space to conduct these experiments. The author would also like to thank the members of BioCurious, with particular thanks to Innokenti Toulokhonov and Irina Toulokhonova, Fabio Rupp, and especially Eric Espinosa for thoughtful discussions and advice throughout the preparation of this manuscript.
Author Disclosure Statement
C.H. is a co-founder of CRISP-HR Therapeutics, which has filed a patent on the underlying CRISP-HR technology (PCT/US2020/012438, WO 2020/146290).
Funding Information
No funding information to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.