
Abstract
Conventional CRISPR approaches for precision genome editing rely on the introduction of DNA double-strand breaks (DSB) and activation of homology-directed repair (HDR), which is inherently genotoxic and inefficient in somatic cells. The development of base editing (BE) systems that edit a target base without requiring generation of DSB or HDR offers an alternative. Here, we describe a novel BE system called Pin-pointTM that recruits a DNA base-modifying enzyme through an RNA aptamer within the gRNA molecule. Pin-point is capable of efficiently modifying base pairs in the human genome with precision and low on-target indel formation. This system can potentially be applied for correcting pathogenic mutations, installing premature stop codons in pathological genes, and introducing other types of genetic changes for basic research and therapeutic development.
Introduction
Gene editing technologies mediated by CRISPR systems provide powerful tools for biotechnology and biomedical research in general.1–3 However, conventional CRISPR technologies rely on the generation of DNA double-strand break (DSBs) at target sites, which may have oncogenic liability. Moreover, for a precision repair, conventional CRISPR technologies requires the activation of homology-directed repair (HDR), which is low in nondividing somatic cells.4,5 Thus, there is a need for the development of new gene editing systems that limit the introduction of DNA DSBs and facilitate precise gene editing without the need for HDR.
To overcome the limitations associated with the requirement of DSB and HDR posed by conventional CRISPR-Cas systems, an elegant gene editing method called base editing (BE) was developed. 6 BE harnesses the DNA targeting ability of a nuclease defective Cas9 (dCas9 or nCas9), combined with the DNA editing activity of the cytidine deaminases APOBEC-1. 6 By directly fusing the deaminase effector to the nuclease-deficient Cas9 protein, these tools introduce targeted point mutations in genomic DNA 6 or RNA 7 without requiring DSBs or HDR activity, and has been successfully applied to models from animals to crops,8,9 highlighting its versatility and biotechnological and biomedical potential. A variation to direct fusion of a deaminase to dCas9/nCas9 has been published, where an effector deaminase is recruited to dCas9/nCas9 through the use of protein–protein interaction domains. 10
An alternative to fusing an effector deaminase to Cas9 is to engineer the guide RNA (gRNA) component of the CRISPR-Cas9 complex as an anchor for recruitment. In this approach, the gRNA is engineered to include an RNA aptamer, which interacts with its cognate ligand fused to an effector protein. This mode of recruitment has been utilized for transcriptional regulation,11,12 the labeling of genomic loci,13,14 and targeted hypermutation.15,16 The separation of the DNA recognition module from the effector module and the use of RNA aptamer for effector recruitment has potential advantages, such as allowing convenient reconfiguration of the system by the mix and match of individual components and simultaneous recruitment of different effectors to different target sites.
Here, we report the engineering and optimization of an RNA aptamer-mediated BE system named Pin-point™. Our data demonstrate that like published direct protein fusion BE systems, Pin-point can edit target cytidines in endogenous sites with high efficiency, low on-target indel formation rate, and low off-target activity. By harnessing orthologous RNA aptamer–RNA binding protein pairings to recruit different DNA deaminases, heterologous BE at separate loci can be conveniently achieved.
Methods
Bacterial experiments
All bacterial experiments, including strains used and specific experimental conditions, are described in the Supplementary Information.
Plasmids
Bacterial and mammalian expression plasmid construction and cloning are described in the Supplementary Information.
gRNA design
Targeting gRNAs were designed manually on SnapGene Viewer (GSL Biotech). All gRNAs used in this study are described in Supplementary Tables S1, S3, and S4.
Cell culture
HEK293T cells were purchased from ATCC (CRL-3216). Cells were grown and maintained at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (Thermo Fisher Scientific), supplemented with 10% fetal bovine serum, 1 × glutamine (Thermo Fisher Scientific), and 1 × antibiotic-antimycotic (Thermo Fisher Scientific).
BE treatments and quantification
For lipofectamine-based transfections (Thermo Fisher Scientific), HEK293T and its derivatives nf2.16 or 293_GFP cells were plated on six-well plates the day before experiments (3.5 × 105 cells/well). Transfections were performed on cells at 75–85% confluence, with a total of 2 μg of a combination of DNA from effector/Cas9 constructs and gRNA constructs at a 3:1 ratio, respectively. Transfections by electroporation were performed on a Neon Transfection System (Thermo Fisher Scientific), according to the manufacturer's instructions. For electroporation, 2 × 105 cells were transfected with 1 μg of a combination of DNA from effector/Cas9 constructs and gRNA constructs at a 3:1 ratio, respectively. All experiments were performed with three biologically independent replicates, analyzed by Sanger sequencing, and quantified by the EditR analytical tool. 17 One sample from each triplicate treatment was analyzed by next-generation sequencing (NGS). Specific BE experiments in mammalian cells are described in the Supplementary Information.
Whole-exome sequencing analysis
Whole-exome sequencing (WES) was carried out by Genewiz. The WES libraries were constructed using Agilent SureSelect Human All Exon (V6 r2) library prep kit and sequenced using Illumina HiSeq with the pair-end 2 × 150 bp format. For details, see the Supplementary Information.
NGS analysis
The primer sequences used in this study are summarized in Supplementary Table S6. All polymerase chain reaction (PCR) amplifications were performed with high-fidelity Phusion Hot Start DNA Polymerase (New England Biolabs), as per the manufacturer's instructions. PCR products were purified with a QIAquick PCR Purification Kit (Qiagen) and submitted to Genewiz for high-throughput sequencing analysis. For details, see the Supplementary Information.
Results
Design of the Pin-point BE system
The Pin-point system consists of three structural modules, as illustrated in Figure 1A and B: (1) a nuclease-deficient Cas9 (dCas9 or nCas9); (2) a programmable chimeric RNA scaffold containing a gRNA (for targeting [2.1] and Cas9 binding [2.2]) and a recruiting RNA aptamer (for effector recruitment [2.3]); and (3) an effector module consisting of a cytidine deaminase ([3.1]) fused to an RNA binding protein [3.2] that specifically interacts with the RNA aptamer. Our prototype system consisted of bacterial vectors expressing a catalytically dead Cas9 protein (dCas9D10A/H840A), an RNA aptamer derived from the operator stem-loop of bacteriophage MS2 (MS2) fused to the 3′ end of the gRNA scaffold, and human activation-induced cytidine deaminase (AID) fused to MS2 coat protein (MCP), which interacts with MS2 aptamer (Supplementary Fig. S1).
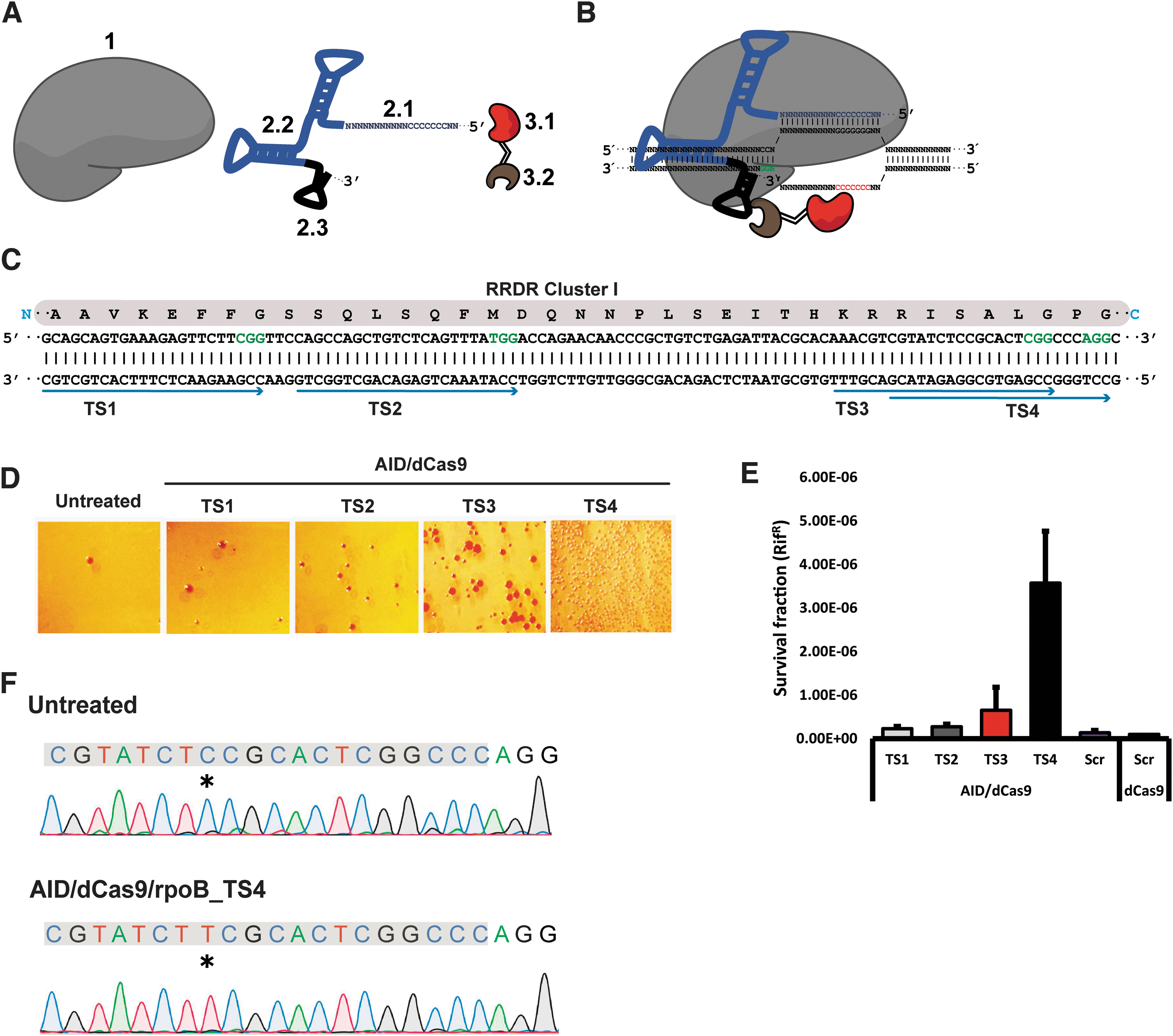
Description of Pin-point™ base editing (BE) platform and proof of principle in prokaryotic cells.
Proof of concept in prokaryotic cells
We used a negative selection approach with the antibiotic rifampicin to test our system in bacteria. Well-defined mutations along the rifampicin resistance determining region (RRDR) of the rpoB gene are associated with rifampicin resistance (Fig. 1C). 18 The RRDR was targeted with four chimeric gRNAs expressing one MS2 motif (rpoB_TS1–TS4; Fig. 1C and Supplementary Table S1) and dCas9. The system expressing AID_MCP and dCas9 is noted as AID/dCas9. Treatment with AID/dCas9 guided by TS4_1 × MS2 resulted in a survival fraction 35-fold higher than cells treated with a scramble gRNA (Fig. 1D and E). Sequence analysis of isolated colonies treated with AID/dCas9/TS4_1 × MS2 revealed that the system introduced a targeted C → T mutation at codon 531, changing a serine to a phenylalanine, a mutation known to induce rifampicin resistance18,19 (Fig. 1F). The higher efficiency observed in TS4_1 × MS2-treated cells might be due to the position of the target C within the protospacer, which in this case sits at position 8 from the 5′ end of the protospacer (i.e., protospacer adjacent motif [PAM] distal). TS2 and TS3 sites have target Cs at positions 12 and 14, respectively, suggesting that distal sites from the PAM sequence within the protospacer region are favored for BE using AID/dCas9 in prokaryotes. Taken together, these data show that a targeted nucleotide modification using an RNA aptamer-based effector recruitment mechanism is a feasible approach for targeted BE.
In order to increase editing efficiency of our prototype system, we sought to engineer each individual module systematically, targeting the rpoB gene with TS4 gRNA for comparison. Switching the Cas9 module from dCas9 to nickase Cas9D10A or Cas9H840A resulted in a 4.6- and 1.7-fold increase in efficiency compared to AID/dCas9, respectively (Supplementary Fig. S2A). Clones treated with AID/Cas9H840A showed random mutations outside the protospacer at a high frequency, a mutational pattern not observed in AID/Cas9D10A (Supplementary Fig. S3A). Next, doubling the number of RNA aptamer sequences (2 × MS2) resulted in a substantial 360-fold increase in the number of surviving colonies compared to scramble-treated cells, and increased the surviving colonies 16-fold compared to AID/dCas9-treated cells (Supplementary Fig. S2A
Finally, we tested rat APOBEC1 and human APOBEC3G as Pin-point effectors. APOBEC1/Cas9D10A showed the highest conversion efficiency, followed by AID/Cas9D10A (Supplementary Fig. S2C). APOBEC1/Cas9D10A induced a high rate of double mutants, whereas APOBEC3G/Cas9D10A targeted nucleotides outside the protospacer (Supplementary Fig. S3B). Together, these data demonstrate that Pin-point is a robust and flexible BE system capable of editing the prokaryotic genome.
Correction of a loss of function mutation in a green fluorescent protein gene in mammalian cells
Mammalian expression constructs are described in Supplementary Figure S4A. In cells, uracil DNA glycosylase (UNG) initiates the repair of U:G mismatches induced by cytidine deamination.20–22 To enhance nucleotide conversion efficiency at the target sites, we fused a phage derived UNG inhibitor peptide (UGI) 23 to Cas9D10A, thus eliciting local UNG inhibition, a strategy previously shown to enhance efficiency of other BE systems. 6 This version of mammalian expression Pin-point platform is noted as AID/Cas9D10A-v1 (AIDv1). The gRNA construct has two MS2 loops at the 3′ end of the CRISPR scaffold (2 × MS2; Supplementary Fig. S4B).
To test AIDv1, we designed a green fluorescent protein (GFP) reporter that harbors an A → G loss of function (LOF) mutation along the chromophore sequence that results in tyrosine to cysteine mutation at position 66, rendering GFP non-fluorescent (nfEGFP; Fig. 2A). We designed a gRNA targeting the non-template strand (NT) of the mutation region (nfEGFP_NT1; Fig. 2A and Supplementary Table S3).

Correction of a loss of function mutation in human cells.
First, we sought to correct the LOF mutation in extrachromosomal DNA. To this end, we transiently transfected the nfEGFP gene in HEK293T cells together with AIDv1 and nfEGFP_NT1 gRNA (Fig. 2B). High GFP conversion was observed in treated cells (Fig. 2B). Quantification by flow cytometry revealed 52.4% GFP+ cells after AIDv1/nfEGFP_NT1 (Fig. 2B).
To examine whether Pin-point has activity on chromosomal DNA, we stably integrated the nfEGFP gene into HEK293T cells (the resulting cell line was named nf2.16). In this experiment, we also included as positive controls the fourth-generation base editors BE4max 24 and AID-BE4, 25 the BE systems expressing APOBEC1 and AID, respectively. nf2.16 cells treated with AIDv1, AID-BE4, or BE4max and an nfEGFP_NT1 gRNA showed 22.2%, 24.3%, and 7.7% correction efficiency, respectively (Fig. 2C). The lower GFP conversion efficiency observed in BE4max-treated cells is due to the target sequence content. The target C in nfEGFP is in position 3 of the protospacer, which is outside the canonical APOBEC1 activity window (positions 4–8 in published BE systems). 6 Moreover, the target C is part of a GC dinucleotide, a motif known to be unfavorable for APOBEC1.6,26,27 Sequencing of treated cells confirmed this, revealing that AIDv1 and AID-BE4 both efficiently edited the crucial nucleotide (C3) at 51% and 60% C → T editing, respectively, whereas BE4max showed 4% C → T editing at the same position, with 58% of C → T editing at the bystander C8 nucleotide (Fig. 2D). Together, these results indicate that Pin-point can edit extrachromosomal and chromosomal sequences in mammalian cells with high efficiency.
BE at endogenous target sequences
To determine the ability of our system to modify endogenous loci in the human genome, we targeted sequences commonly known as Site 2, Site 3, and Site 4 in HEK293T cells (the sequences and genomic coordinates of these sites are described in Supplementary Table S4), which have been previously extensively studied by conventional nuclease-dependent CRISPR genome editing28,29 and by other BE systems. 6 In addition to AID as effector, we constructed an APOBEC1-expressing system, designated APOBEC1/Cas9D10Av1 (A1v1). We investigated the on-target efficacy and on-target indel formation rates of AIDv1 and A1v1 at these sites. Sequence analysis revealed that both AIDv1 and A1v1 resulted in efficient C → T conversion with high purity at all three sites (Fig. 3A–C and Supplementary Figs. S5–S7). These observations show that our modular approach can effectively target endogenous genomic sequences. The different activity profiles observed between AIDv1 and A1v1 is likely due to the specific sequence preference of each deaminase tested. The preferred motif for AID is WRC (W: A/T, R: A/G) and YC for APOBEC1 (Y: T or C), 26 and such preferences have been reported by others characterizing different BE effectors in direct fusion BE systems. 27 Since BE with a type II Cas nuclease is constrained by PAM availability and the relative position of the target nucleotide within the protospacer, using deaminases with different editing profiles could be advantageous for expanding the range of target genes that can be modified using a base editor.

Pin-point-mediated BE at endogenous loci. HEK293T cells were electroporated with the indicated constructs and subjected to Sanger sequencing, and the BE outcomes were measured using the EditR analytical tool.
17
First-generation AIDv1 and A1v1 constructs were targeted to Site 2
To examine indel formation induced by AIDv1 and A1v1, we analyzed the frequencies of these events at Sites 2, 3, and 4 in edited and unedited cells. Indels in edited cells were detected at a low frequency and approximately two orders of magnitude lower than conventional wild-type Cas9 (wtCas9) at these sites (Supplementary Figs. S5–S7). Indels induced by AIDv1 and A1v1 are comparable to the levels of indels reported for other BE systems.6,30 These results indicate that our first-generation Pin-point base editors can efficiently edit endogenous genomic loci and generate indels at levels similar to other base editor systems and substantially below those of conventional CRISPR-Cas systems.
Codon optimization and increased UNG local inhibition
To improve Pin-point, we codon optimized the protein coding genes, and added additional copies of UGI and nuclear localization signal to the Cas9 module (Supplementary Fig. S4C). These modifications are similar to the optimization of the BE system.24,27 The upgraded Pin-point constructs are noted as v2 (i.e., AIDv2 or A1v2). We tested the impact of these changes on BE efficiency and on-target indel formation in HEK293T Site 2 and Site 4. Both AIDv2 and A1v2 resulted in efficient BE at these sites (Fig. 3D and E and Supplementary Figs. S8 and S9). Inclusion of AID-BE4 and BE4max as positive controls, targeted with aptamer-less gRNAs, indicated that all the constructs induced low levels of indels with similar distributions, although subtle differences in indel frequencies were evident between the base editors at Site 2 (Supplementary Figs. S8 and S9). Interestingly, there is a marked reduction in indel formation from AIDv1 and A1v1 to AIDv2 and A1v2, likely due to the increased presence of UGI in our second-generation constructs (Supplementary Figs. S5A and B and S8A and B). Note that the reduction in indel formation does not impact BE efficiency. Together, these data show that our optimized second-generation Pin-point base editors exhibit higher efficacy compared to our first-generation v1 system, while inducing lower on-target indel formation rates at these target sites.
BE by heterologous effectors at separate loci using different RNA aptamers
An important feature of the Pin-point system is its flexibility and modular design. Pin-point can potentially deploy heterologous BE effectors to separate genomic loci by harnessing different RNA aptamers to recruit distinct effector modules fused to cognate RNA aptamer binding proteins (Fig. 3F), without the need of delivering multiple large orthogonal Cas9 molecules. 31 This dual-aptamer BE differs from the published dual BE systems in which simultaneous cytidine and adenosine BE occur within the same protospacer.32,33 As a proof of principle, we tested a Pin-point system for dual-aptamer cytidine BE at both Site 2 and Site 4 simultaneously with different deaminases. To this end, we fused AID to the PP7 coat protein (PCP), a functional ortholog of MCP that binds to PP7 aptamers, 11 and called it AIDv2PCP. This fusion protein can be recruited to a target site via gRNAs containing two PP7 aptamers in the same configuration as the MS2-harboring gRNAs described before.
For this set of experiments, we engineered our constructs to express each module from a separate plasmid (Supplementary Fig. S4D). HEK293T cells edited by either AIDv2PCP/PP7 gRNA targeting Site 2 or A1v2/MS2 gRNA targeting Site 4 individually showed robust BE at each site (Fig. 3G and Supplementary Fig. S10A and B). AIDv2PCP/PP7gRNA generated a similar BE profile compared to AIDv2/MS2gRNA (Fig. 3D and G). We tested editing Sites 2 and 4 simultaneously by transfecting both the AIDv2PCP/PP7gRNA (Site 2) pair and the A1v2/MS2gRNA (Site 4) pair into HEK293T cells. Dual-aptamer BE was detected, with each site showing the distinct signature editing profile of AID and APOBEC1, respectively (Fig. 3H and Supplementary Fig. S10C). The results provide an example that Pin-point is capable of performing dual-aptamer BE simultaneously at different genomic loci with different effectors.
Initial off-target assessment
In order to estimate the CRISPR-dependent off-target activity of our second-generation systems initially, we looked at selected known off-target sites of Site 2, Site 3, and Site 4 (summarized in Supplementary Table S5), which were previously identified by chromatin immunoprecipitation of dCas928 and by the GUIDE-seq method, 29 and which have been analyzed with other base editors. 6 High-throughput sequencing revealed undetectable off-target editing at the off-target sites of Sites 2 and 3 (Supplementary Figs. S11 and S12). Off-target editing at S4O1, a Site 4 off-target site, was detected with all base editors tested, while only A1v2 and BE4max induced detectable C → T editing at S4O2 (Supplementary Fig. S13). With these limited set of off-target sites, our initial studies indicate, as expected, that both Pin-point and BE systems exhibit fewer off-target effects (fewer off-target sites and lower activity at the off-target sites where detectable editing is present) than the conventional CRISPR-Cas9 gene editing.
To assess the CRISPR-independent mutational footprint of Pin-point initially, we subjected HEK293T cells edited at Site 2 with AIDv2 (or BE4max as a positive control) to exome-wide sequencing analysis. At the cell population level, the data revealed no significant change in global SNP distribution (Supplementary Fig. 14A) or C → T mutations at known hot spots (Supplementary Fig. 14B). It is worth noting that BE4max was reported to induce detectable CRISPR-independent C → T mutations when clonal cell populations were analyzed34,35 as opposed to heterogeneously edited cell populations, as examined here. Further studies with higher sensitivity are warranted for determining the exact level of CRISPR-independent mutation effects of Pin-point AIDv2 and A1v2.
Targeted gene disruption by induction of a premature stop codon
BE has been harnessed to induce premature stop codons.36,37 We sought to test the ability of the Pin-point BE platform to induce a stop codon at Q157 in an EGFP reporter gene stably integrated in HEK293T cells (293_GFP cells; Fig. 4A). EGFP was targeted with AIDv2 and an EGFP_TS1 gRNA (Supplementary Table S3). Flow cytometry analysis revealed 25% of GFP– cells in the treated population (Fig. 4B and C). Sequence analysis confirmed that AIDv2 resulted in high efficiency BE at the target site (Fig. 4D and Supplementary Fig. S15A and B).
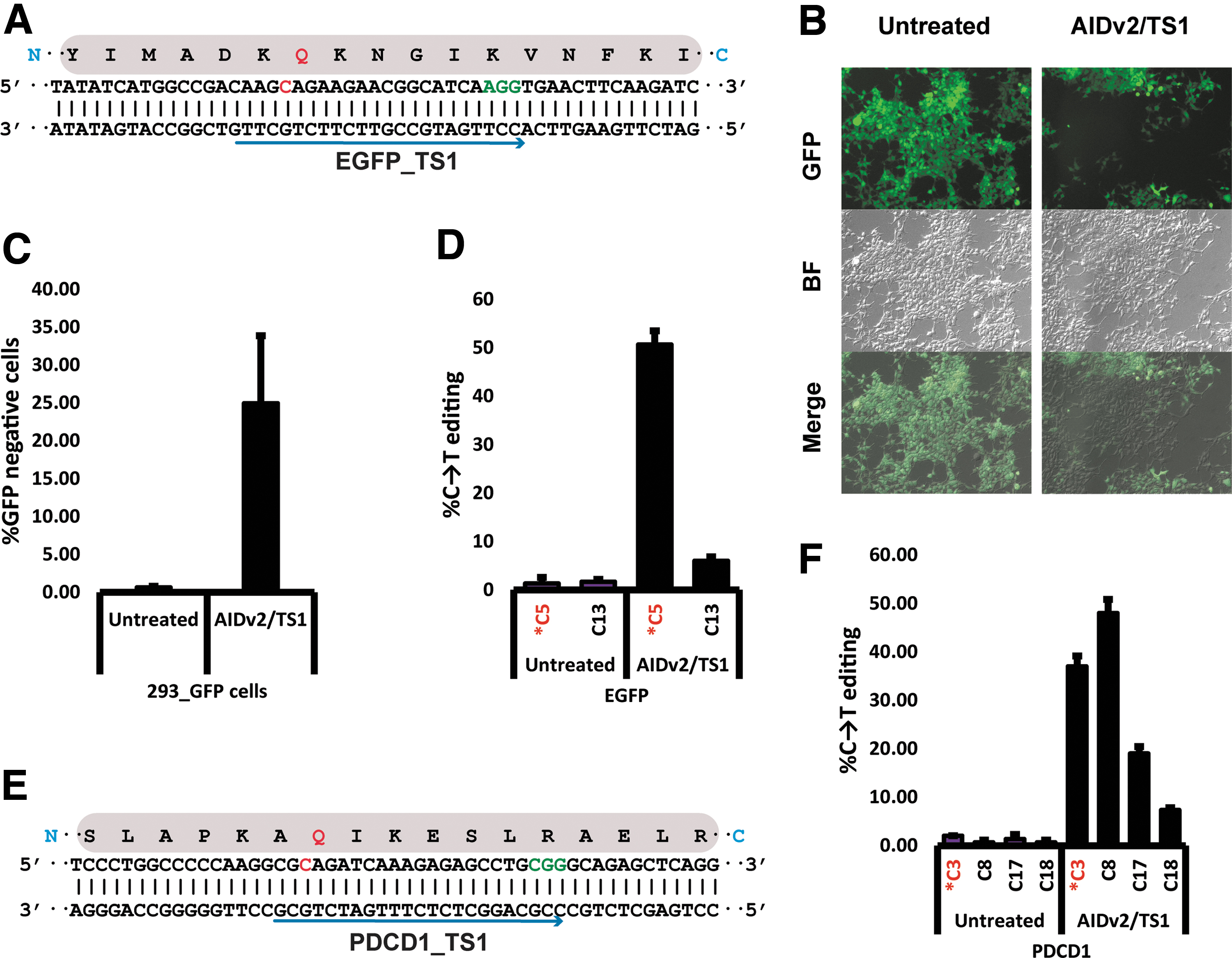
Introduction of a stop codon in a GFP reporter gene and an endogenous site in human cells.
To assess the ability of our system to knockout an endogenous gene, we designed a gRNA targeting codon Q133 of PDCD1, which encodes the immune checkpoint protein PD-138 (Fig. 4E and Supplementary Table S4). AIDv2 resulted in nearly 40% C → T conversion at C3, converting codon Q133 (CAG) to a stop codon (TAG) in K562 cells (Fig. 4F and Supplementary Fig. S15C and D). We observed bystander C editing with similar efficiency at C8, and with lower efficiency at C17 and C18 (Fig. 4F and Supplementary Fig. S15C), where the C8, C17, and C18 editing to T do not change the encoded proteins (all silent mutations). Together, these results provide initial evidence that Pin-point can introduce a nonsense mutation into an endogenous gene.
Discussion
CRISPR technologies provide extraordinary tools for rewriting genetic information, which is expected to have a far-reaching impact on science and technology. One exciting prospect of CRISPR-based technologies is the development of genome editing agents as medicines, which can be used in humans specifically to modify pathogenic or therapeutically relevant genes.39–41 Recently, engineered nuclease- and HDR-independent CRISPR systems have been developed for efficient precision genome modification technologies.6,7,10,42–44 These approaches use a nuclease-deficient Cas9 and gRNA as sequence-specific recognition apparatus to harness non-nuclease DNA modification enzymes, such as cytidine deaminases, DNA methylases, or reverse transcriptases, in order to induce modifications at target loci free from DNA breaks, further enabling CRISPR-based technologies to broad clinical applications.
In this paper, we have explored an alternative strategy for recruiting non-nuclease DNA editing enzymes to the target sequence by the gRNA component—a system we have called Pin-point. This study shows that Pin-point can induce efficient BE at target sites, with low indel formation rates compared to conventional CRISPR gene editing technology. Our second-generation base editors (i.e., AIDv2 and A1v2) exhibit improved editing properties over the first-generation Pin-point editors. Our initial and limited analyses of CRISPR-dependent off-target editing indicates similar levels of off-target editing compared to BE systems and lower off-target rates compared to conventional CRISPR gene editing technology. Our initial analyses of CRISPR-independent off-target effects using WES analysis showed no widespread detectable C → T or G → A editing for AIDv2 or for BE4max when unsorted populations of cells were studied. Notably, BE4max has been reported to induce detectable cytidine deamination in a CRISPR-independent manner when the whole genome of single clones of edited cells is analyzed.34,35 Thus, future studies are required to characterize the CRISPR-dependent and CRISPR-independent off-target effects of Pin-point further. Moreover, the potential off-target effect on RNA should be also characterized, especially for the APOBEC1 Pin-point base editors, since BE4max expressing wild-type APOBEC1 has been found to edit the transcriptome in a CRISPR-independent manner. 42
An appealing feature of Pin-point is its modular design: the functionality of DNA recognition and effector action reside in separate molecules, and the interaction of the two functional modules is encoded by a chimeric gRNA molecule that can be easily altered. This feature is potentially important for a number of applications. For example, as the BE efficiency at different genetic loci varies, optimization of base editor configuration is usually needed. With Pin-point, the CRISPR protein module and the effector module can be individually engineered and/or optimized, and a large repertoire of Pin-point constructs can be conveniently constructed by the mix and match of individual components for achieving optimal gene editing at a desired target site. Moreover, with Pin-point, BE by heterologous editors at separate loci can be conveniently achieved (Fig. 3F–H). By application of multiple pairs of orthologous RNA aptamers and their cognate RNA-binding proteins, Pin-point could use a single nCas9 protein to recruit heterologous effectors, such as next-generation cytosine base editors with reduced off-target deamination, 35 engineered base editors with narrower activity window for highly precise BE, 45 adenine base editors,24,46 or other DNA interacting effectors to target different loci simultaneously with the desired effectors without cross-reactions between the heterologous effectors. This application could be important when multiplex BE by heterologous effectors is preferable, such as in next-generation chimeric antigen receptor T-cell engineering, where the modification of multiple genes is called for.
In addition to Pin-point, other CRISPR systems have been engineered using gRNA components and RNA aptamers for recruiting effectors.11,12 For example, CRISPR-X, recruits a hyperactive variant of AID by an RNA aptamer localized at the tetraloop and stem loop 2 of gRNA.15,16 However, unlike Pin-point, CRISPR-X generates DNA hypermutations over a wide range around and beyond the target protospacer sequence at low frequency. As a result of this, CRISPR-X has been used for protein evolution and for generating antibody diversity.15,16 The distinct DNA modification patterns between CRISPR-X and Pin-point are likely to stem from the use of a hyperactive form of AID in CRISPR-X and other design differences between the two systems.
In summary, we have engineered an RNA aptamer-mediated BE system. Our data indicate that this system is effective for BE with low on-target indel formation. With a modular design that fully separates the DNA modification module from the DNA recognition module, which enables facile modifications of these components, Pin-point provides an engaging addition to existing BE technologies. Devoid of the requirements of DNA DSB and HDR, this system and other BE systems are set to provide powerful tools for genetic engineering and for therapeutic development.
Footnotes
Acknowledgments
The authors thank Dr. Nicola McCarthy for carefully editing the manuscript and for discussion, and Drs. Tatiana Litvin-Vechnyak, Shan Wan, Jon Moore, Chris Lowe, Anja Smith, and Amanda Smith for support and discussions. Thanks also to Dr. Husam Al-Hraishawi for generating the cell line nf2.16. We gratefully acknowledge access to the HPC facilities and support of the computational STEM and bioinformatics scientists from the Office of Advanced Research Computing (OARC) at Rutgers University.
Author Disclosure Statement
S.J. and J.C.C. have filed patent applications through Rutgers University describing the technology presented in this work. T.S., J.J.L., J.A.H., J.S., and C.M.W. are employees at Horizon Discovery, which has an exclusive license for therapeutic, diagnostics and service applications of the Pin-point™ base editing system.
Funding Information
Research in this study was supported by the National Institutes of Health under award number US NIH/NCI R21 CA216604 (S.J.); by Fulbright—SENESCYT (Instituto Ecuatoriano de Fomento del Talento Humano; grant number 15130816; J.C.C.); by Rutgers University—Biomedical Sciences Fellowship (J.C.C); by Rutgers University TechAdvance Research Grant (Project 205483; S.J., J.C.C.); by the National Institute of General Medical Sciences (NIGMS; grant NIH T32 GM008339; V.T.); and by funding provided by Horizon Discovery.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.