
Abstract
We describe CALITAS, a CRISPR-Cas-aware aligner and integrated off-target search algorithm. CALITAS uses a modified and CRISPR-tuned version of the Needleman–Wunsch algorithm. It supports an unlimited number of mismatches and gaps and allows protospacer adjacent motif (PAM) mismatches or PAMless searches. CALITAS also includes an exhaustive search routine to scan genomes and genome variants provided with a standard Variant Call Format file. By default, CALITAS returns a single best alignment for a given off-target site, which is a significant improvement compared to other off-target algorithms, and it enables off-targets to be referenced directly using alignment coordinates. We validate and compare CALITAS using a selected set of target sites, as well as experimentally derived specificity data sets. In summary, CALITAS is a new tool for precise and relevant alignments and identification of candidate off-target sites across a genome. We believe it is the state of the art for CRISPR-Cas specificity assessments.
Introduction
CRISPR-Cas-based medicines are being developed to treat serious diseases, and their safety evaluations must include specificity assessments. Early studies showed that genomic sites with mismatches with respect to the sequence of the variable portion of the CRISPR RNA molecule (guide RNA [gRNA]) can also be cut1–4 and edited, and potentially create deleterious mutations. These are referred to as off-targets. There are a variety of empirical methods to evaluate CRISPR-Cas specificity, including cellular methods, such as GUIDE-seq, 5 and fully reconstituted biochemical methods, such as Digenome-seq. 6
To complement experimental off-target discovery methods, in silico off-target searching is a commonly used tool to identify regions in the genome similar to the gRNA. Genomic sites that have a similar sequence to the gRNA and contain an appropriate protospacer adjacent motif (PAM) may be inadvertently cut and edited by CRISPR-Cas in an in vivo environment. In silico off-target discovery is used to compile lists of candidate off-target sites, which are then interrogated by targeted amplicon sequencing methods such as rhAMP-seq. 7 However, current in silico candidate off-target identification methods fail to take into account several important factors in the behavior of gRNA binding to potential cut sites. Experimentally, it has been observed that off-target sites can have not only mismatches with respect to the gRNA but also gaps in the alignment, resulting in what are known as DNA or RNA bulges. 8 Also, due to the constant description and creation of novel Cas variants,9–11 whose PAM sequence is not always fully characterized, it becomes useful to have in silico methods with flexible PAM selection. It is thus important that in silico off-target searching methods can incorporate multiple gaps in their searches, as well as allowing multiple PAMs, PAM mismatches, or even PAMless searches.
A survey of existing tools for in silico off-target search shows several common shortcomings. Tools such as E-CRISP, 12 CHOPCHOP,13,14 or CRISPOR 15 rely on alignment algorithms developed for next-generation sequencing applications: bowtie2, 16 bowtie, 17 and bwa, 18 respectively. None of these algorithms are optimized for alignments with multiple mismatches and gaps (MMGs) across the relatively short sequences of a typical gRNA (∼18–25 bases). These tools also have limitations in the maximum number of mismatches and do not allow for the inclusion of gaps in their searches. Other tools such as Cas-OFFinder 19 or CRISPRitz 20 are designed for CRISPR applications and allow for the inclusion of an unlimited number of gaps. However, they use a brute force index and match approach that scales exponentially as more gaps are added in the search. Cas-OFFinder and CRISPRitz also have the limitation of adding gaps only on either the DNA or the RNA, but not on both. In addition, Cas-OFFinder or CRISPRitz return all possible alignments for effectively the same site, requiring further processing steps to select unique off-targets from redundant sites. In one, not uncommon, example, when alignments with up to three MMGs were requested, more than 2.3 million different alignments were returned for the same guide, but only about 400 were unique sites up to three MMGs. This redundancy grows as the number of MMGs increases, producing large files and making further operations difficult. Most tools, with the exception of Cas-OFFinder, do not allow PAMless or flexible PAM searches. Finally, native inclusion of genomic variants via a standard Variant Call Format (VCF) file is not included in many tools, with the exception of CRISPOR (although only for the on-target site) and CRISPRitz.
We have developed a CRISPR-Cas-aware ALigner for In silico off-TArget Search (CALITAS). CALITAS implements a modified semi-global version of the Needleman–Wunsch (NW) algorithm, 21 with parameters set specifically for CRISPR off-target searching. It allows (1) unlimited mismatches, (2) unlimited mixtures of gRNA and DNA gaps, (3) gaps between guide and PAM, (4) differential scoring for gRNA and DNA gaps, and (5) mismatches within the PAM. Scoring penalties have been tuned to ensure mismatches, DNA gaps, and RNA gaps have the same effective net cost for gRNA-length alignments while maintaining a preference for, in order: mismatches, RNA gaps, and then DNA gaps. Finally, the algorithm produces a single “best” alignment at any position within a target sequence with the ability to suppress overlapping alignments, enabling standardization and clear threshold-based selection of candidate off-targets using a simple count of mismatches plus gaps.
CALITAS natively supports exhaustive searches across a genome of interest or gRNA alignments to a given list of sites (e.g., the output of a Digenome-seq experiment). Alternate variant alleles from the 1000 Genomes Project, or similar data sets, can be directly incorporated into a search via user-provided VCF files. A typical gRNA run against the human reference genome plus all 1000 Genomes variants with an allele frequency >1% completes in about an hour on a modern processor using 10 threads and can be scaled on a per guide basis using cloud environments such as AWS Batch. We used CALITAS to identify candidate off-targets for 41 gRNAs that have PAMs for SpCas9, SaCas9, and AsCas12a. We compared our predictions with Cas-OFFinder and CRISPRitz and showed that CALITAS identifies more candidate sites, likely because multiple gaps are not being aligned optimally in other algorithms. Finally, we used CALITAS to align gRNAs to experimentally detected sites and to show that the inclusion of gaps is biologically relevant.
Methods
Description of the CALITAS algorithm
The core of CALITAS is the semi-global alignment algorithm that aligns the query (gRNA) to one or more local regions of a target sequence. Alignment is performed twice: once to the positive strand sequence and once to the negative strand sequence.
Alignment matrices are constructed for the gRNA, without the PAM, versus the target sequence, as with NW, but with a modified scoring system that uses different gap penalties, depending on whether the gap occurs in the query or target. A user-specified cutoff for differences (mismatches plus gap bases) in the gRNA is translated into a minimum alignment score, and the matrix searched for cells that (1) represent full alignments of the query and (2) have scores meeting the threshold (see Fig. 1). All such cells are back-traced and filtered to ensure they do not exceed the differences cutoff in order to produce candidate alignments.

Key CALITAS parameters and alignment scores.
If one or more PAM(s) are specified, the alignment proceeds to an extension phase (see Fig. 2). Each candidate alignment is extended by (1) allowing up to a user-defined number of gaps between the gRNA and PAM and (2) aligning each PAM sequence with mismatches but without gaps. The highest scoring extension per candidate alignment is selected and, if all thresholds are met, returned.
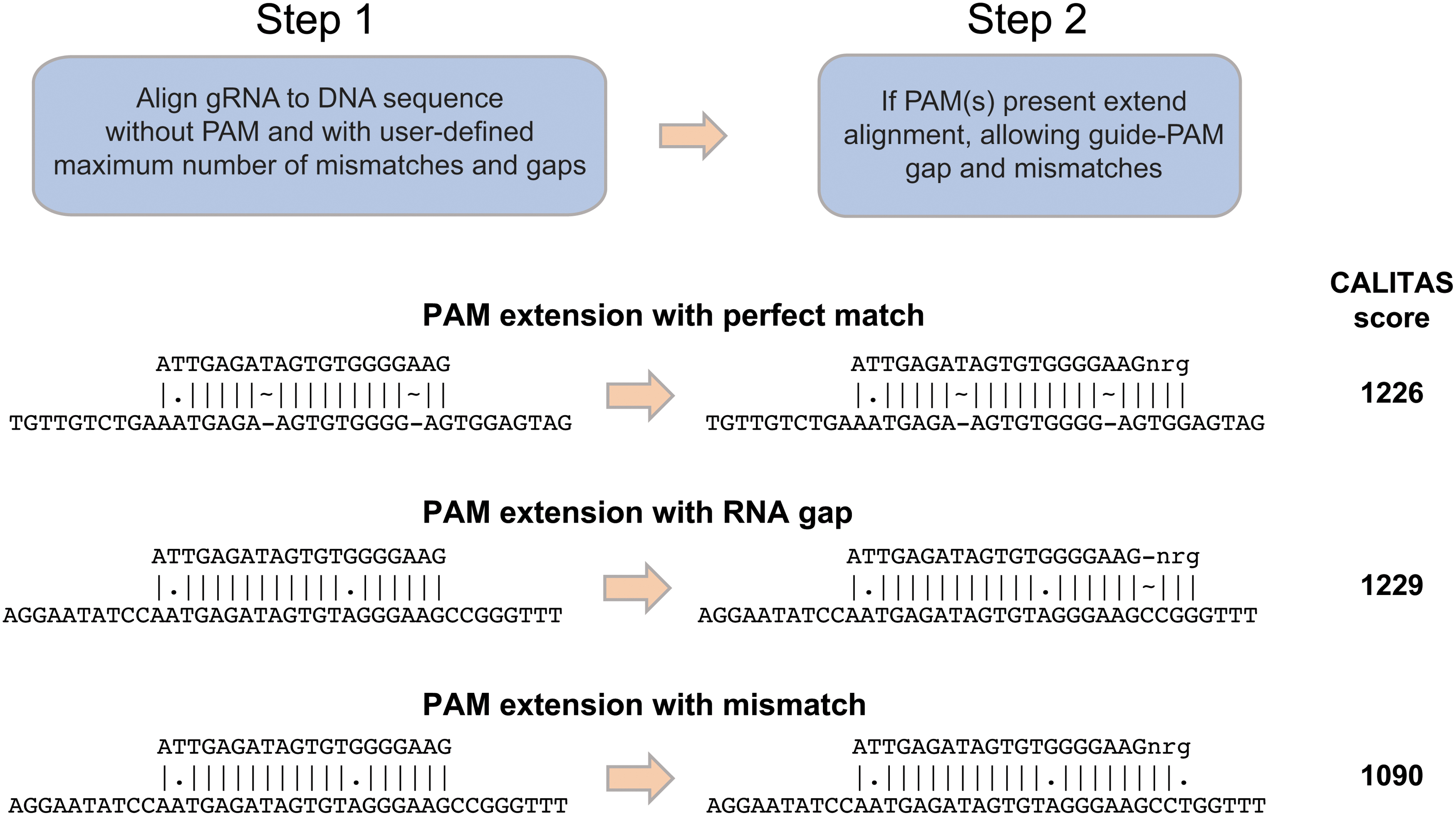
Description of CALITAS's two-step approach to align gRNA and protospacer adjacent motif (PAM) to a target genomic DNA sequence. Three examples of PAM extension alignments are shown, including perfect matches and MMGs. For each example, the resulting alignments after steps 1 and 2 are shown, together with their final CALITAS score.
CALITAS reverse complements query and/or target sequences during alignment in order to ensure that any PAM(s) present are located at the end of the query sequence during alignment. As a result, the terminal cells in the candidate alignment matrixes always represent the PAM-adjacent base from the gRNA. Since each terminal cell represents an alignment terminating at a different target base, CALITAS can back-trace the single best-scoring alignment per terminal cell, instead of all acceptable alignments per terminal cell. This produces a single candidate alignment per possible PAM location, reducing both the computational complexity and the number of redundant alignments produced. Furthermore, a max overlap parameter controls the reporting of overlapping alignments to reduce further the number of redundant alignments reported. If two acceptable alignments overlap on the target sequence by more than max overlap base pairs, only the higher scoring of the two alignments is reported. Optionally, multiple alignments per site that fall within user-defined constraints can be returned (Supplementary Fig. S1).
While internally CALITAS uses a standard scoring system involving penalties for mismatches, gap opens, and gap extends, it accepts a set of scoring parameters that are more intuitive to biologists expressed as net costs. Net costs are accepted for (1) mismatches in the gRNA, (2) mismatches in the PAM, (3) gaps in the DNA, and (4) gaps in the RNA. The net cost is the total change in alignment score for the event; for example, for a mismatch, the net cost is decomposed into the cost of subtracting one match plus the score of adding one mismatch.
CALITAS has two modes: SearchReference for genome searches and AlignToReference to align a gRNA to a list of sites. In SearchReference, the core aligner is used to identify candidate off-target sites in a genome by windowing over the genome, aligning the guide sequence to small sections (default: 1,000 bp) of the target genome. Windowing ensures that the memory requirements of CALITAS remain low while enabling parallelization across multiple CPU cores. Windows are overlapped sufficiently to ensure that no alignments are missed at window boundaries, and redundant alignments found due to the overlapping are removed prior to output. The genome search also accepts a VCF of variants to integrate into the genome during the search. CALITAS generates all possible haplotypes by clustering variants that are close enough to one another to be covered by a single alignment and then inserting each possible combinations of alleles into the target sequence. Allele frequency of these synthetic haplotypes is calculated as the minimum allele frequency present in the synthetic haplotype (Supplementary Fig. S2). This approach represents the worst-case scenario for all haplotypes and was used rather than inferring or mining data sources for haplotype frequencies, which will be lower and many excluded in the genome. That said, users should note the allele frequency of these synthetic haplotypes should be examined and redetermined. In AlignToReference, given a list of sites, a gRNA alignment is performed for the sequences surrounding each site.
Alignment coordinates returned by CALITAS are 0-based half-open, as is used in the standard bed file format.
Reference files used for alignments
The human genome reference hg38, GRCh38, was downloaded from the Golden Path resource: http://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/. The file, hg38.fa.gz, with a last modified date of January 15, 2014, 21:14, and size of 938 Mb was downloaded. All chromosome (contigs), after being extracted, were used in the alignments.
For comparison with the GUIDE-seq alignments in Tsai et al., 5 we used the genome reference hg19: http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/. The version used is the UCSC release based off the NCBI GRCh37 patch 13 from February 2009. In addition to the main chromosomal sequences, hg19 contains mitochondrial sequences, unplaced sequences, and alternate haplotypes.
Human common variants were derived from the 1000 Genomes Project (https://www.internationalgenome.org/data). Phase 3 from the 1000 Genomes Project was used in this study, which contains sequencing data from 2,504 individuals, representing 26 populations and containing more than 84 million catalogued variants.
Variant calls in VCF format were downloaded from the European Bioinformatics Institute (EBI) site: http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/supporting/GRCh38_positions/. Files that matched the pattern “*.sites*.vcf.gz” where “*” indicates any character(s), were retrieved from that location.
The files were further processed using the PrepareVcf tool included in the aligner to: (1) standardize chromosome names, (2) filter out variants or alleles with a frequency <1.0%, and (3) remove extraneous data in the “INFO” field to reduce the file size. This yielded a single VCF file containing the variants to be used with CALITAS.
Digenome-seq data and results for RSQ5138
Digenome experiments were performed as in Maeder et al. 22 We used the gRNA RSQ5138 (ATTGAGATAGTGTGGGGAAG) complexed to SpCas9 protein that targets a region in the promoter of HBG1 and HBG2. The final list of 987 Digenome sites was obtained using the pipeline at https://github.com/editasmedicine/digenomitas and a template score threshold of 15 (Supplementary File S1).
41 gRNAs used with simultaneous PAMs for AsCas12a, SaCas9, and SpCas9
To design the 41 gRNAs that were used in the comparison, we searched for sites with the sequence pattern TTTVNNNNNNNNNNNNNNNNNNNNNGGRRT, which stands for gRNAs of a length of 20 bp, with PAMs of AsCas12a (TTTV), SpCas9 (NGG), and SaCas9 (NNGRRT), in autosomes of hg38 (excluding sex chromosomes, mitochondrial chromosome, alt, and random sequences). Next, we removed sites where the identical sequence occurred more than once in the genome or contained a homopolymer of length ≥3 in the target sequence. Finally, we filtered out sites with GC content outside of the window 35–70% or located <500 bp away from repetitive elements. From this final list, we randomly selected 41 gRNAs. The sequence for the 41 gRNAs can be found in Supplementary File S1.
CALITAS alignments to randomized sites in the genome
To characterize the null distribution of the alignments returned by CALITAS, we aligned the 41 gRNAs to 100,000 random sites in hg38. The random sites were generated using the bedtools random command 23 (v2.28.0), subtracting regions from repeat masker, centromere, and gaps. Alignments were made using permissive PAMs for AsCas12a (TTTN), SaCas12a (NNGRRN), and SpCas9 (NRG), as well as a PAMless search.
Comparison with Cas-OFFinder and CRISPRitz
CRISPRitz 20 v2.1.1 (350fc90) was run using docker. First, hg38 was indexed to support up to three bulges using the command index-genome with the option -bMax 3. Then, the search command was used with the options -mm 3 -bDNA 3 -bRNA 3 -t -th 10. Note that this version returns coordinates which are neither 0-based nor 1-based (see multiple alignments in Supplementary Fig. S3).
Cas-OFFinder 19 v2.4 (a521753) was run using the wrapper to add bulges from https://github.com/hyugel/cas-offinder-bulge.git (b5d6ab4). To compare with CALITAS and CRISPRitz, Cas-OFFinder was run using all available CPUs, although it is worth noting that Cas-OFFinder can be run using GPUs as well. The search parameters specified a maximum of three mismatches and three bulges.
To deduplicate output files from CRISPRitz and Cas-OFFinder, we first grouped alignments using bedtools cluster 23 (v2.28.0), and the best alignment from each cluster was selected using pandas groupby 24 (v0.23.4).
To compare running times, we used a machine with an Intel i7-9750H CPU @ 2.60 GHz processor, 16 GB 2,400 MHz DDR4 RAM, running macOS Catalina v10.15.4. A table of running times for all the aligners using different options is given in Supplementary Figure S5.
To compare the number of sites up to three MMGs for all the aligners, we created an area-proportional Euler diagram using eulerAPE v325 (Fig. 4B).
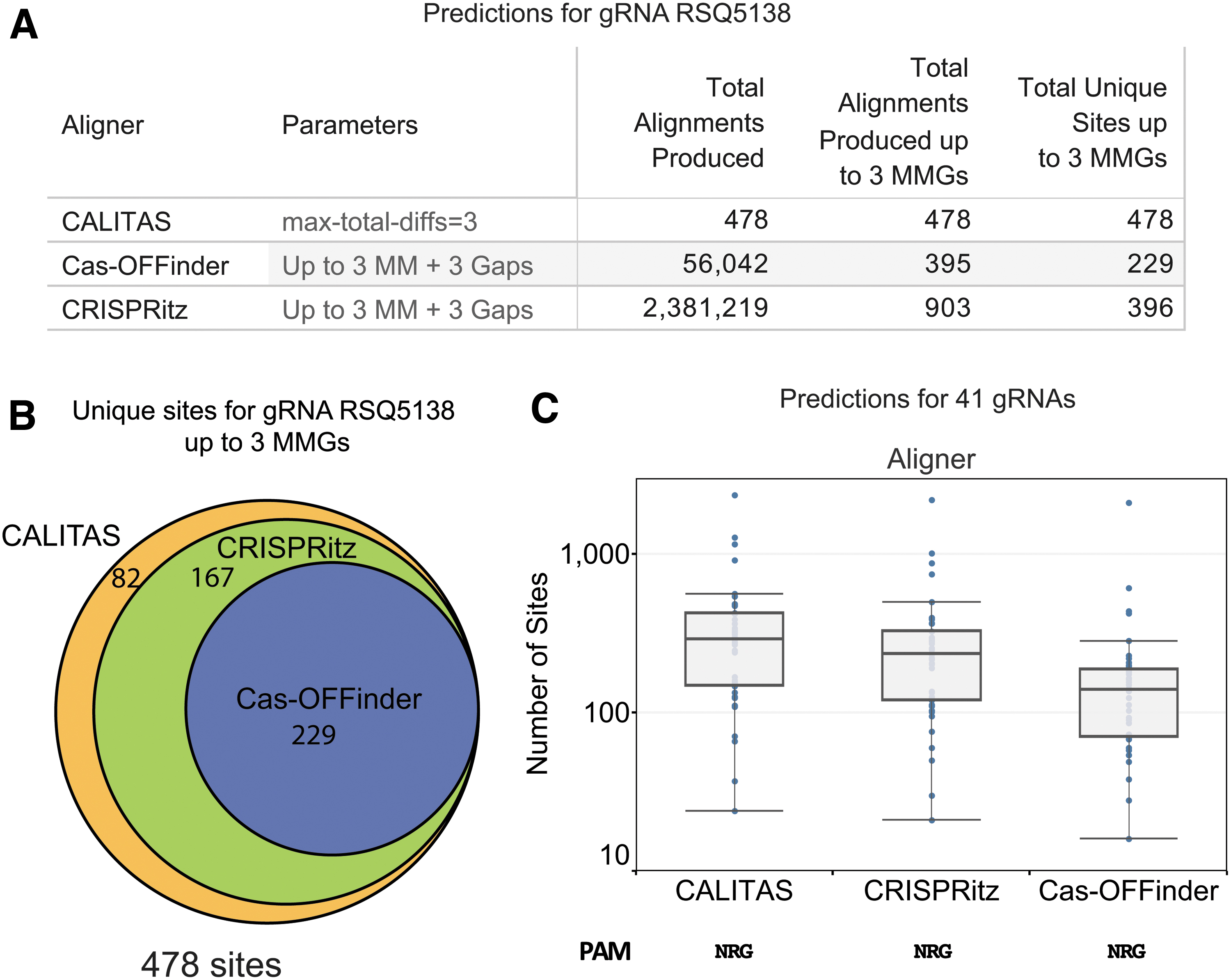
Comparison of the predictions of CALITAS, CRISPRitz, and Cas-OFFinder.
Comparison of CALITAS-identified sites to CHANGE-seq sites
To compare CALITAS alignments against the experimentally detected CHANGE-seq sites reported in Lazzarotto et al., 26 we ran CALITAS in SearchReference mode for the 110 guides with reported off-targets using the NRG PAM. To compare the CHANGE-seq sites with the sites identified by CALITAS, the ∼100,000 CHANGE-seq sites were realigned to their respective guides using the CALITAS AlignToReference command, generating files and alignments that made it easy to compare between CALITAS and CHANGE-seq.
Comparison of CALITAS-identified sites to GUIDE-seq sites
To compare CALITAS alignments against the gap-less GUIDE-seq alignments present in Tsai et al., 5 four gRNAs were selected—EMX1, HEK293 sgRNA4, VEGFA site2, and VEGFA site3—with a total of 362 sites in the original publication. We excluded seven sites from further consideration, since they had different sequences than in the hg19 reference genome used in our analysis. Unfortunately, we were unable to find an explanation for these differences or locate a reference genome which matched the target sequences. The remaining 355 sites were submitted for alignment with CALITAS using the AlignToReference mode. Given the 20 bp query sequence, PAM (NGG), chromosome, and start position of the query, the CALITAS output was then compared in a base-by-base alignment against the original gap-less GUIDE-seq alignment. The parameters used for CALITAS alignment were the following: maximum allowed gaps between the guide sequence and PAM, 3; maximum allowed mismatches in the PAM, as CALITAS does not allow gaps in the PAM, 1; maximum allowed difference (total MMGs) between the guide and genome, 5. Mismatches in the pam region are penalized more than twice a mismatch in the query sequence (−260 vs. −120). Furthermore, alignments with at least 10 bp overlapping were considered redundant and removed.
Results
To illustrate CALITAS candidate off-target identification capabilities using its SearchReference mode, it was run on a set of 41 gRNAs that were 20 nt in length that had simultaneous perfect PAMs for the nucleases AsCas12a, SaCas9, and SpCas9 (see Methods and Fig. 3A and B). It is important to note that we chose these 20 nt guides for illustrative purposes, but CALITAS can be run for guides of any length. For the 41 gRNAs and the TTTN PAM for AsCas12a, CALITAS identified a median of 53 sites with three or fewer MMGs, including the on-target (min = 5, max = 327). The list obtained for the SaCas9 PAM NNGRRN had markedly more sites (median = 146, min = 15, max = 921). Using the NRG PAM for SpCas9, we obtained a median of 288, a minimum of 24, and a maximum of 2,356 sites. Finally, we also identified off-targets for the 41 gRNAs when no PAM was used, yielding a median of 1,519, a minimum of 127, and a maximum of 8,113 sites. CALITAS predicts that AsCas12a has much fewer off-target sites than SaCas9 or SpCas9—a result in line with the expected prevalence of their PAMs in random double-stranded sequence: 1 in 32, 1 in 8, and 1 in 4, respectively.
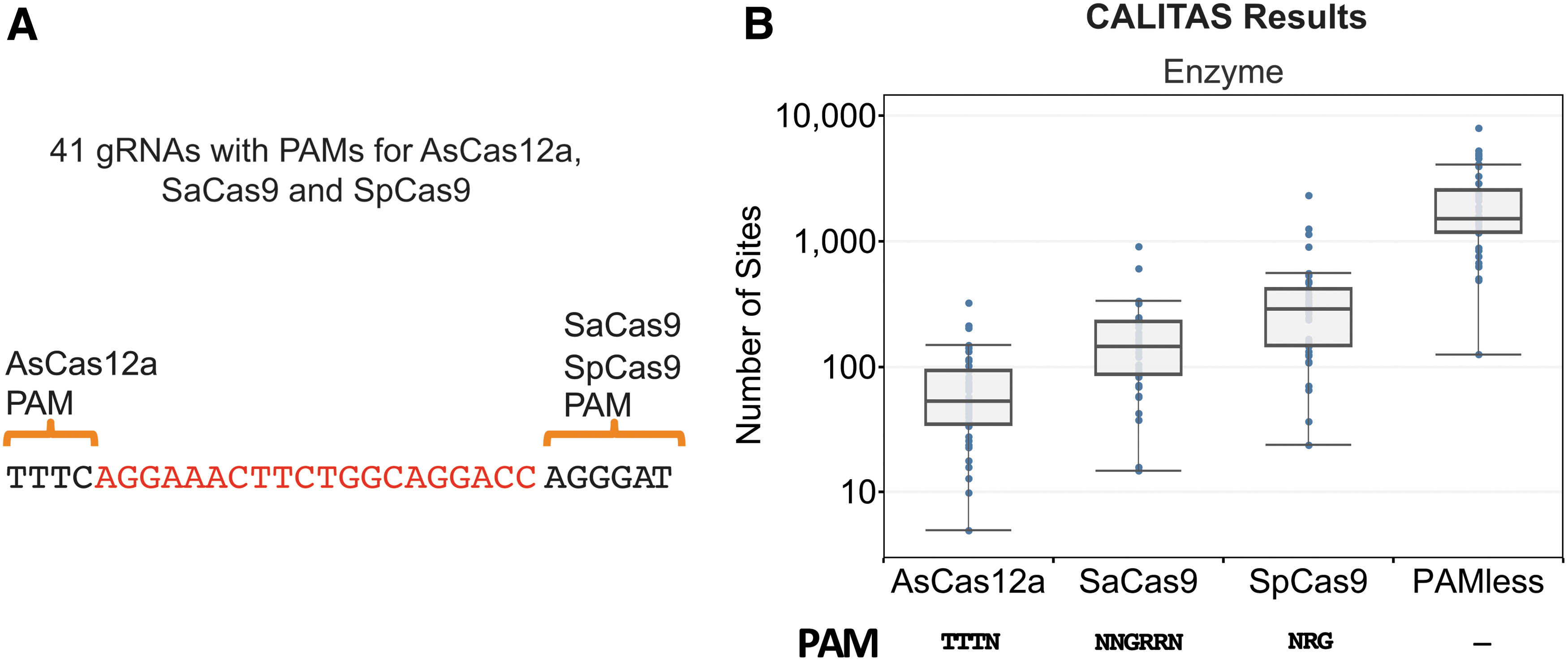
Off-target candidates for 41 guides.
To understand the null distribution of the sites with three MMGs that CALITAS found, we selected a set of 100,000 random locations throughout the genome and, using CALITAS AlignToReference mode (see Methods), aligned all 41 guides using the PAMs for AsCas12a, SaCas9, SpCas9 PAMs, as well as no PAM. The results in Supplementary Figure S4 show that random sites have predominantly between 8 and 10 mismatches, and fewer than 6.3E–6 are expected to have fewer than three MMGs, with AsCas12a being the more specific enzyme.
Next, CALITAS was compared to two of the leading tools: Cas-OFFinder and CRISPRitz (see Supplementary Fig. S5 for a comparison of running times using the guide RSQ5138 and various settings). One of shortcomings of Cas-OFFinder and CRISPRitz is that they return most sites multiple times, each with a different alignment. As an example, unlike CALITAS that returns one alignment per site, when using CRISPRitz, a single site was returned with 40 alignments (Supplementary Fig. S3). This results in large files with many redundant sites (see Fig. 4A). To make a meaningful comparison, we deduplicated their result files (see Methods) and selected the best alignment per locus (i.e., with the lowest number of MMGs). Comparison of the alignments up to three MMGs (Fig. 4B) showed that Cas-OFFinder was the tool that returned fewer sites, with 229 alignments. All those alignments plus 167 more were found by CRISPRitz. CALITAS was the tool that returned most sites, including the previous 396 plus 82 novel sites not found by Cas-OFFinder or CRISPRitz. Examination of those sites revealed that 17 contained PAM mismatches and 65 contained mixtures of DNA and RNA gaps, which are not considered in those tools and explained the greater number of sites found by CALITAS.
Next, we used Cas-OFFinder and CRISPRitz to make predictions for the same 41 guides that had PAM for various Cas enzymes. At the time of writing this article, we were unable to use CRISPRitz for AsCas12a or SaCas9, and thus we only used SpCas9 in our comparisons (see Fig. 4C for their results up to three MMGs). For CRISPRitz, we found a median of 234, a minimum of 21 and a maximum of 2,197 sites. For Cas-OFFinder, the 41 gRNAs had a median of 139, a minimum of 16 and a maximum of 2,109 sites. In all 41 cases, the number of candidate off-target sites found by CALITAS is larger. This is likely due to better handling of gaps within the alignments and the inclusion of mismatches in the PAM.
To compare sites identified by CALITAS with actual CRISPR experimental data, we obtained Digenome-seq data for a guide that displayed many off-targets (Methods). This SpCas9 guide (RSQ5138) had a total of 987 Digenome-seq off-targets. We use the AlignToReference mode of CALITAS (see Methods) that allows the user to obtain gRNA alignments to a given set of genomic locations. To understand the importance of including multiple gaps, as CALITAS does by default, we also ran CALITAS with parameters so that gaps were prevented in the alignments. In Figure 5A, we show the alignments to the 987 sites using CALITAS default parameters (with gaps) and without gaps. The distribution of MMGs using CALITAS with gaps had an average of 4.4 MMGs (green line in Fig. 5A). On the other hand, forcing the alignments to not have gaps resulted in alignments with an average of 6.6 MMGs (red line in Fig. 5A). To illustrate the reason for this discrepancy, we show two examples of alignments with CALITAS default parameters (Fig. 5B) and without gaps (Fig. 5C). Allowing multiple gaps results in an alignment with one mismatch and two gaps (DNA gaps in this case), whereas the alignment without gaps results in an alignment with 10 mismatches. It is important to note that a high number of CALITAS alignments had more than one gap: 316 had two gaps and 123 had three gaps (Supplementary Fig. S6 and Supplementary File S1). From these results and analysis, it is clear that supporting multiple gaps, as CALITAS does, is an important feature for CRISPR-Cas-based aligners.
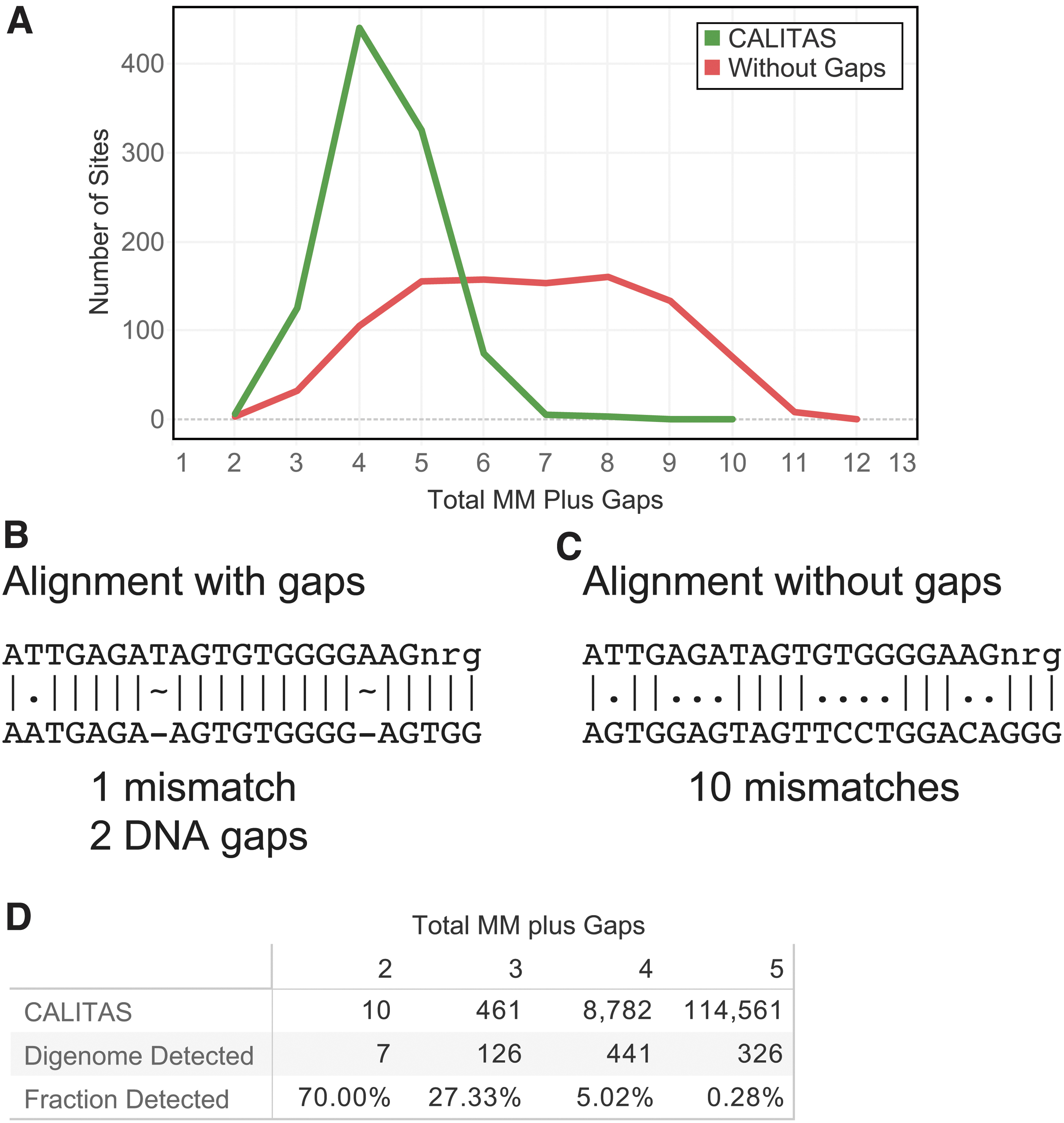
CALITAS-identified off-target sites and comparison with Digenome-seq sites for the guide RSQ5138.
To illustrate the relationship between CALITAS-identified and empirically determined off-targets better, we performed a comparison between sites identified by CALITAS and the ∼200,000 sites for 110 SpCas9 guides appearing in the CHANGE-seq publication by Lazzarotto et al. 26 As shown in Supplementary Figure S7, the comparison shows that CALITAS-identified sites with three MMGs (a median of 252 sites) are validated by CHANGE-seq 18% of the time. That value decreases to 3% for the sites with four MMGs (a median of 4,944 sites). On the other hand, CALITAS can be very useful to identify candidate off-target sites that have very few MMGs, even though they are not detected in a CHANGE-seq experiment. For example, 50% of the CALITAS-identified sites with two MMGs are not found in the CHANGE-seq experiments in Lazzarotto et al. 26 Given that these sites have only two MMGs, they would warrant further evaluation and verification with tools such as targeted amplicon sequencing.
Finally, in order to explore further the importance of adding gaps to make better off-target predictions, we reanalyzed a previous data set from the original GUIDE-seq paper, 5 which contained alignments including only mismatches, without gaps. We selected four guides with multiple off-targets (between 16 and 146) and used CALITAS to align the guides to the off -target sites using the same PAM, NGG (Fig. 6). CALITAS obtained alignments that had overall a lower number of MMGs than the original GUIDE-seq alignments, which had no gaps. Although the differences were small for some of the guides, for guides such as VEGFA site 2, the differences were very significant, again highlighting the importance of adding gaps when predicting off-targets.
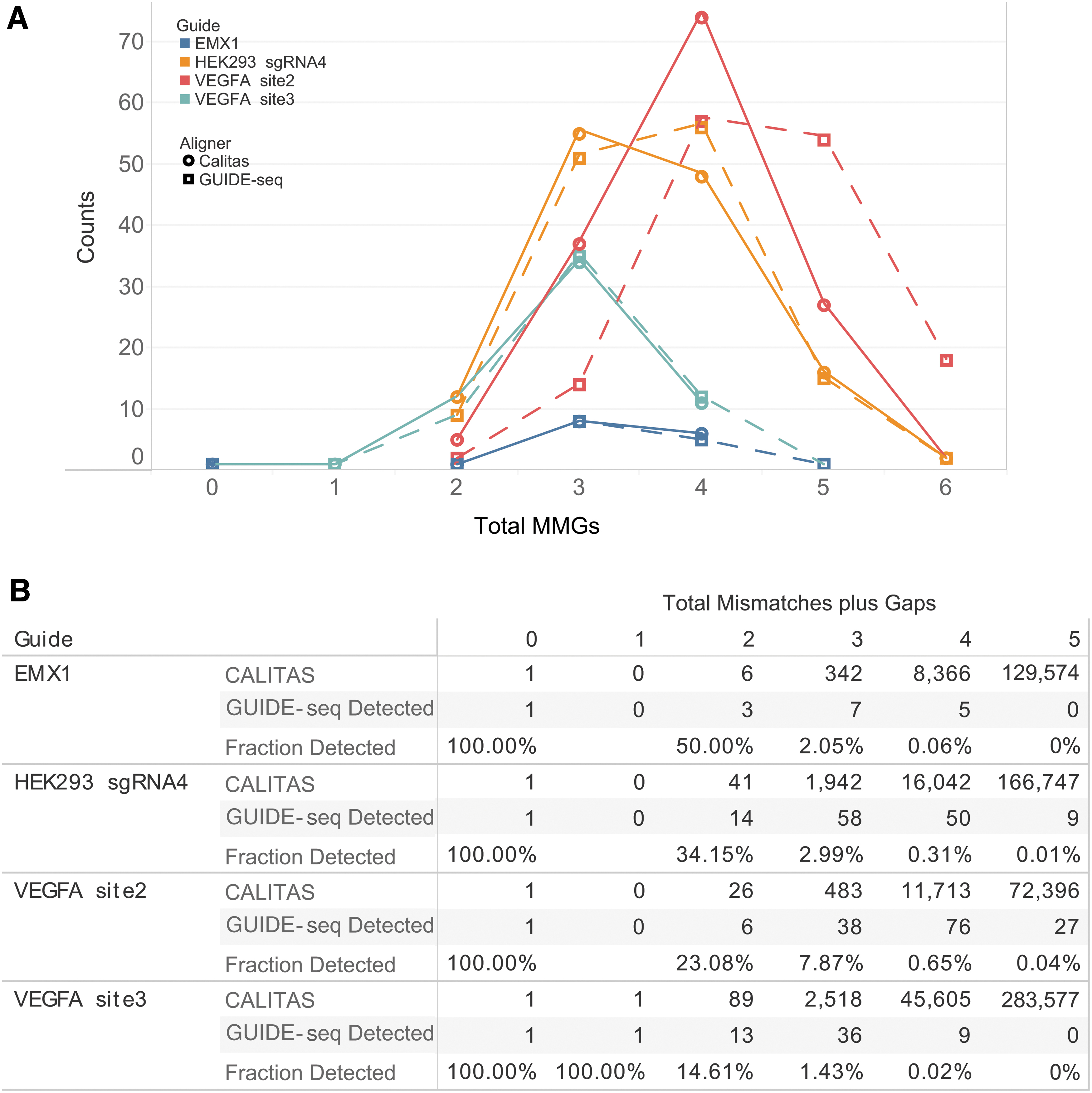
CALITAS-identified off-target sites and comparison with GUIDE-seq sites for the guides EMX1, HEK293 sgRNA4, VEGFA site2, and VEGFA site3 in Tsai et al.
5
From these examples, it is evident that many of the sites identified by CALITAS with a high number of MMGs are not validated experimentally (Figs. 5D and 6A). As can be seen in Figure 5D, ∼27% of the CALITAS-identified candidate sites with three MMGs are detected in Digenome-seq, whereas only ∼5% of the CALITAS-identified sites with four MMGs are detected. For the in-cells GUIDE-seq sites in Figure 6B, CALITAS-identified sites with three MMGs are detected between 1.4% and 7.9%, and only between 0.02% and 0.7% of the sites with four MMGs.
Discussion
Here, we present CALITAS, an integrated CRISPR-Cas-aware aligner and search tool for local alignments and genome searches. CALITAS was originally developed to enable a single aligner to be used for genome-wide searches as well as for local alignment to experimentally derived sites. In the development process, it was clear that producing a single best alignment per site was critical to achieving this goal. With CALITAS, it is now possible to align sites from a GUIDE-seq experiment and directly compare that site based solely on coordinates to a genome-wide search, knowing that the coordinate will align directly without use of additional tools (e.g., BedTools Cluster). In the process, several other beneficial features were incorporated, such as the incorporation of variants, improved scalability for multi-gapped alignments, and better PAM handling.
CALITAS is optimized for finding alignments with multiple gaps in a reference using its SearchReference mode, returning the single best alignment for each off-target site. CALITAS also offers PAM flexibility by allowing PAM mismatches and being able to run with multiple PAMs or PAMless. CALITAS off-target searches can also easily include genomic variants (e.g., from the 1000 Genomes Project) by accepting a standard VCF file as input. All of these make CALITAS a tool appropriate for generating candidate off-target lists.
CALITAS can also be used with its AlignToReference mode to align a guide and PAM or PAMs to a list of sites specified by their coordinates. This is useful to align a guide to candidate off-target sites obtained experimentally using a method such as Digenome-seq or GUIDE-seq. This alignment is made using the same NW algorithm used in SearchReference, and it ensures that in silico and experimental sites use the same alignment scheme.
We show that CALITAS more closely mimics CRISPR-Cas biology by allowing the incorporation of multiple gaps, with reduced biases between gRNA and DNA gaps. This behavior is easily controlled by user-defined net cost parameters. For example, PAM mismatches can be eliminated by setting a very negative PAM-mismatch net cost (e.g., −10,000). By default, CALITAS uses parameters such that there is a small preference (relative penalty differences slightly >0.8%) for mismatches over gRNA gaps and over DNA gaps. This facilitates breaking ties between alignments while ensuring that their penalties are very similar. We gave a small preference for gRNA over DNA gaps, since alignments with the former have one more base pair. Comparison with Digenome-seq data showed that many experimentally detected off-targets have alignments with multiple gaps (145 with three or more gaps; Supplementary Fig. S2). This highlights the importance of using off-target searching tools that can efficiently incorporate multiple gaps.
Comparison of CALITAS-identified candidate off-targets with experimentally observed sites in Digenome-seq, CHANGE-seq, and GUIDE-seq showed that 25.3% and 1–8% of the in silico identified sites up to three MMGs are found, compared to only 5% and <1%, respectively, of the sites up to four MMGs. Since the number of in silico sites grows exponentially with the number of mismatches, this suggests that verification panels only return a significant fraction of true positive off-targets for in silico sites up to three MMGs
In a workflow where off-target sequencing panels are quickly made after guide selection using CALITAS, we would recommend selecting sites with three or fewer MMGs. This represents a balance of testing obvious sites with between zero and two MMGs and sites with three MMGs where there is a good likelihood (8–25% in these studies) that they will have some experimental evidence as off-targets. CALITAS-identified sites with four MMGs or more have a lower likelihood (<1–5%) of being found with experimental methods, and the number of candidate sites grows exponentially to a point that targeted deep sequencing is no longer straightforward or even feasible. This workflow consists of a first round of targeted sequencing using in silico identified CALITAS off-target sites with three or fewer MMGs, followed by a second round of targeted sequencing using wet-lab detected off-target sites with four or more MMGs (using GUIDE-seq, Digenome-seq, CHANGE-seq, or similar methods). A workflow like this is an optimization for quickly identifying and verifying off-target activity for selected guides while using orthogonal methods to search and verify off-target editing activity comprehensively.
Conclusion
CALITAS is a state-of-the-art CRISPR-Cas-aware aligner and search tool that can be easily run on any computer running Java. The software is available at https://github.com/editasmedicine/calitas
Footnotes
Acknowledgments
We thank Mike Dinsmore for assistance in reviewing the code.
Authorship Disclosure Statement
T.F. is a current employee of Fulcrum Genomics. T.W., G.G., D.Z, C.J.W., and E.M. are current employees and shareholders of Editas Medicine. M.I. is a former employee and shareholder of Editas Medicine and was employed by Editas at the time this work was conducted.
Funding Information
No funding was received for this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.