
Abstract
CRISPR-Cas9 mediated genome editing methods are being used for the analysis of gene function. However, it is hard to identify gene knockout mutants for genes whose knockout does not cause distinct phenotypes. To overcome this issue in the disease vector, Aedes aegypti, a transgenic Cas9/single guide RNA (sgRNA) method, was used to knock out the eye marker gene, kynurenine 3-monooxygenase (kmo), and the juvenile hormone receptor, Methoprene-tolerant (Met). PiggyBac transformation vectors were prepared to express sgRNAs targeting kmo and Met under the control of the U6 promoter. Transgenic Ae. aegypti expressing kmo-sgRNA or Met-sgRNA under the control of the U6 promoter and enhanced green fluorescent protein (eGFP) under the control of the hr5ie1 promoter were produced. The U6-sgRNA adults were mated with AAEL010097-Cas9 adults. The progeny were screened, and the insects expressing eGFP and DsRed were selected and evaluated for mutations in target genes. About 77% and 78% of the progeny that were positive for both eGFP and DsRed in kmo-sgRNA and Met-sgRNA groups, respectively, showed mutations in their target genes.
Introduction
The yellow fever mosquito, Aedes aegypti, transmits several deadly viruses, including yellow fever, Zika, dengue and chikungunya, and causes deaths all over the world every year.1,2 The viruses are transmitted to humans through the bite of an infected female Aedes mosquito, which mainly acquires the virus while feeding on the blood of an infected person. 3 RNA interference (RNAi) has been widely used as a powerful tool for in vivo gene functions analysis in insects. 4 However, RNAi is not highly efficient in mosquito larvae due to problems associated with double-stranded RNA (dsRNA) stability and delivery to the site of action. 5 The CRISPR-Cas9 system has been widely used as an alternative approach to study gene function.6,7 Normally, single guide RNA (sgRNA) and Cas9 mRNA/protein are injected into embryos, and the progeny are screened for mutants. 6 This method is both labor- and time-intensive, since it takes three to five generations to generate homozygous lines. 8 Knocking out some target genes can induce lethality in homozygous individuals. The heterozygous individuals are maintained and crossed to obtain homozygous individuals for in vivo functional studies. For some target genes, the knockout may not produce any distinct phenotype. In this case, enzyme assays or cloning and sequencing need to be performed to identify mutations.9,10 The fluorescent markers can be used for selection by site-specific knock-in at the target site. 11 In mosquitoes, the efficiency of CRISPR-Cas9-mediated knock-in is relatively low. 12 Also, this method takes a long time to generate homozygous lines, 13 and this method is also not suitable for lethal genes.14,15
Previous studies have used multiple sgRNAs for achieving knockout in one generation in mammals 16 and insects.17,18–21 By using the multiple sgRNAs strategies, the loss-of-function mutants could be obtained in one generation. Still, for the target genes with no detectable phenotype, the T7 endonuclease 1 (T7E1) assay or sequencing needs to be performed to identify mutations. We used single marker gene sgRNA mixed with multiple-target gene sgRNAs to identify the loss-of-function mutants in Ae. aegypti. This method increased the frequency of loss-of-function phenotypes in G0. 18 A recent study has shown that the use of transgenic Ae. aegypti expressing Cas9 increased gene-editing efficiency. 22 To extend the toolkit of CRISPR-Cas9 system in the mosquitoes, we produced transgenic Ae. aegypti expressing kmo-sgRNA or Met-sgRNA under the control of the U6 promoter and enhanced green fluorescent protein (eGFP) under the control of the hr5ie1 promoter. The sgRNA/eGFP-expressing mosquitoes were crossed with those expressing Cas9 and DsRed (the AAEL010097-Cas9 line 22 ). The DsRed and eGFP markers were used to identify progeny expressing both Cas9 and sgRNA, which contained mutations for target genes.
Methods
Plasmid construction
The Gibson Assembly (New England Biolabs, Ipswich, MA) method was used for preparing U6-sgRNA constructs. The design and synthesis of sgRNA targeting the kynurenine 3-monooxygenase (kmo) gene, which codes for an enzyme involved in eye pigmentation, was reported previously. 18 To construct the U6-kmo-sgRNA vector, the sgRNA and the promoter of ubiquitous U6 snRNA (AAEL017774) 23 were cloned into piggyBac transformation (pBac-hr5ie1-egfp) vector. 24 A 1,001 bp region at the 5′ end and a 1,301 bp region at the 3′ end of the U6 snRNA were amplified using gene-specific primers (Table 1) and cloned into pBac-hr5ie1-egfp vector. A double-stranded sgRNA scaffold was generated using two unique oligonucleotides (Table 1), and it was cloned into the middle of U6 snRNA promoter vector. Plasmid DNA was extracted using the Plasmid Plus Midi Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. The transposase mRNA was prepared using the mMESSAGE mMACHINE T7 Transcription Kit (Thermo Fisher Scientific, Waltham, MA) with XhoI (New England Biolabs) linearized IFP2 vector as a template. 24
Insect rearing and microinjection
The Ae. aegypti AAEL010097-Cas9 and Liverpool IB12 (LVPIB12) strains were maintained at 27 ± 2°C with a photoperiod of 14 h light:10 h dark and 60–70% relative humidity. The mosquito larvae were fed on bovine liver powder and maintained in water at 26°C. Pupae were sexed by size and kept in separate containers. After emergence, the adults were fed on 10% sucrose solution. For egg collection, around 200 females and 200 males were transferred to a cage for mating. To induce oviposition at 4 days after mating, the females were artificially fed on defibrinated sheep blood. The mosquito embryonic microinjections were performed following the method described previously. 25 Briefly, the mosquito eggs were collected 20 min after oviposition, aligned on a glass slide, and then microinjected. After injection, the eggs were kept at 26°C. On the third day after injection, they were dropped into water for hatching. The hatched larvae were maintained using the same protocol described above.
U6-sgRNA strain development
The Liverpool IB12 strain was used for genetic transformation. Embryos were microinjected with a solution containing 200 ng/μL U6-sgRNA plasmid and 300 ng/μL transposase mRNA. The injected embryos were hatched on the fourth day. The G0 larvae were maintained and sexed during the pupal stage. After emergence, the adults were mated with opposite sex wild-type Liverpool IB12 strain adults. The G1 progeny were screened under eGFP fluorescent light.
RNA isolation and cDNA synthesis
Total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific). The First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) was used to convert 2 μg RNA to cDNA via reverse transcription. The New England Biolabs 2 × Taq Mix and the sgRNA-specific primers (Table 1) were used to amplify the sgRNA templates.
Mutant screening and analysis
To knock out the target gene, the U6-sgRNA lines were mated with the AAEL010097-Cas9 line. 22 The G1 offspring were screened using GFP and red fluorescent protein fluorescent lights. Only larvae that were positive for green and red fluorescence were selected, and they were maintained in separate containers. The mutant phenotype was screened during the fourth instar larval stages. The genomic DNA isolation was performed using a DNeasy Blood & Tissue Kit (Qiagen) following the manufacturer's instructions. The polymerase chain reaction (PCR) products were purified using a QIAquick PCR Purification Kit (Qiagen) following the manufacturer's instructions. The mutations were verified by T7 endonuclease I (T7E1) assay, and the PCR products were cloned into TA vector and sequenced as described previously. 18 For T7E1 assay, 200 ng PCR products were hybridized in NEBuffer 2 (New England Biolabs) under the following conditions: 95°C for 5 min, 95–85°C at −2°C/s, 85–25°C at −1°C/s, and held at 4°C. Then, the mixtures were digested with 10 IU T7E1 enzyme (New England Biolabs) at 37°C for 15 min. Following digestion, the reaction solution was checked on 2% agarose gels containing Gel Red (Biotium, Fremont, CA). The PCR products were cloned into TOPO pCR4 vector (Thermo Fisher Scientific) and sequenced by the Sanger sequencing method.
Results and Discussion
Expression of kmo-sgRNA by injection of U6-kmo-sgRNA plasmid DNA into embryos that express Cas9 induced mosaic eye phenotype in G0
To test if kmo-sgRNA (Fig. 1A) expressed under the control of the U6 promoter (Fig. 1B) could induce mutations in the target gene, 100 AAEL010097 Cas9 embryos were injected with 200 ng/μL U6-kmo-sgRNA plasmid DNA. Of the 49 hatched larvae, seven showed the mosaic eye phenotype (Fig. 1C). The genomic DNA from the mutants was extracted and used as the template to amplify the fragment of the kmo target site and used in the T7E1 assay. As shown in Figure 1D, the target fragment was digested into two expected fragments. Furthermore, the PCR products were cloned into TA vector and sequenced. Six out of seven clones sequenced showed 3–8 bp deletions (Fig. 1E). These data suggest that the kmo sgRNA expressed under the control of the U6 promoter could induce target-specific mutations, resulting in the mosaic eye phenotype in Ae. aegypti.
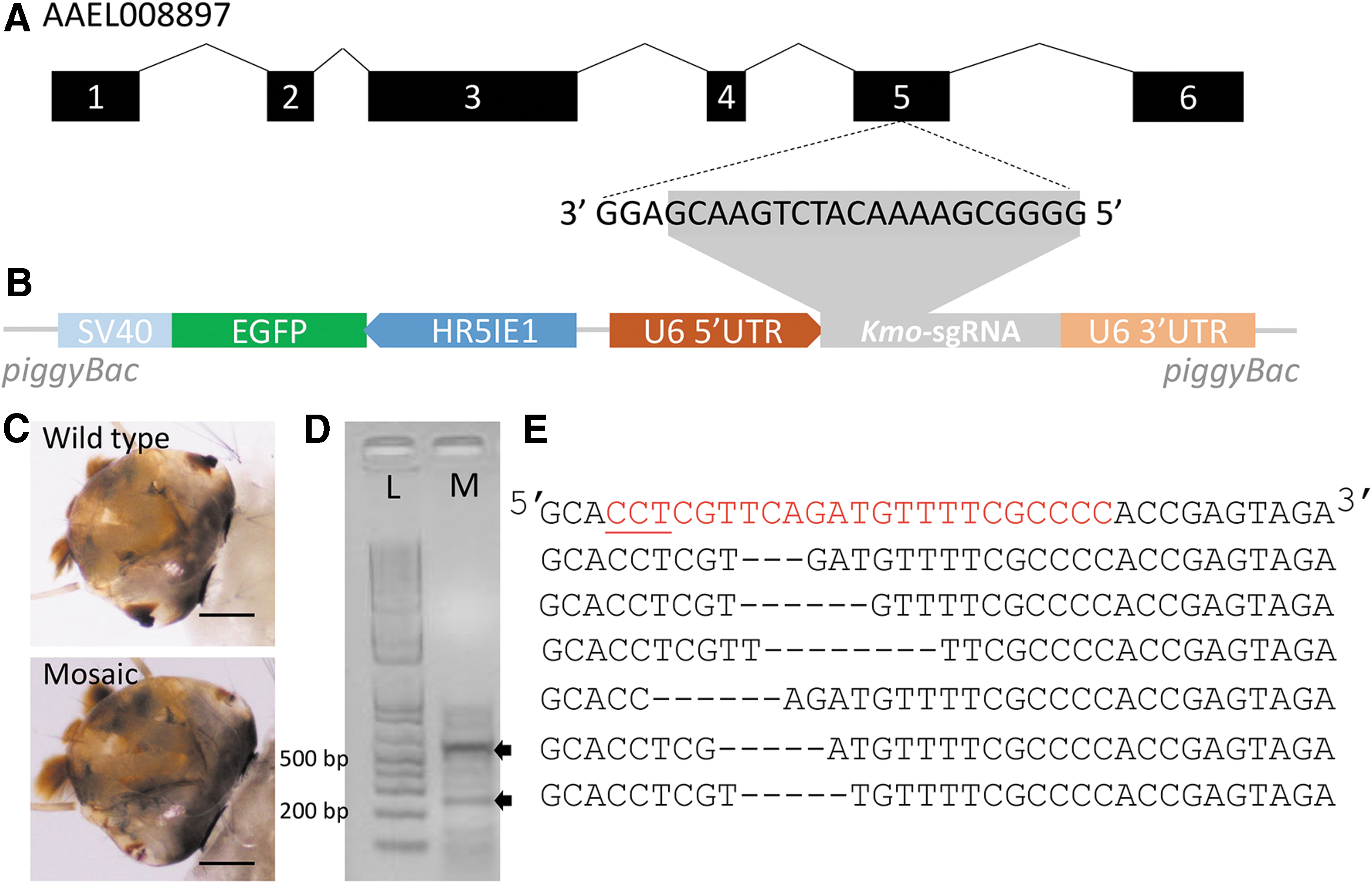
Target-specific mutagenesis of kmo gene and mosaic eye phenotype induced by U6-kmo-sgRNA vector.
Generation of kmo mutants using the transgenic CRISPR-Cas9 system
Three eGFP-positive larvae were identified in the G1 progeny developed from 500 embryos injected with the U6-kmo-sgRNA construct and transposase mRNA. One larva was successfully maintained until the adult stage (male) and mated with 10 Liverpool females. The eggs laid were hatched and maintained until the third instar larval stage and screened under the eGFP fluorescent light. The eGFP-positive larvae (44/150) were selected and kept in separate containers. To enlarge the population, the insects were sexed during the pupal stage and mated with opposite sex Liverpool IB12 adults.
To confirm the expression of sgRNA in vivo, total RNA was isolated from four U6-kmo-sgRNA pupae. RNA (2 μg) was used to synthesize cDNA. The AAEL010097-Cas9 pupae were used as the control. As shown in Supplementary Figure S1, the kmo-sgRNA was detected in U6-kmo-sgRNA insects but not in the Cas9 strain. These data show that sgRNA was successfully expressed in U6-kmo-sgRNA transgenic insects.
The U6-kmo-sgRNA-positive G2 adults were mated with opposite sex AAEL010097-Cas9 adults. The G3 larvae were checked for the presence of eGFP and DsRed during the third instar larval stage. Only the larvae that showed eGFP and DsRed fluorescence were selected and kept in separate containers. When we crossed the AAEL010097-Cas9 line male and the U6-kmo-sgRNA female, none of the larvae developed from this cross showed the mosaic eye phenotype. (Fig. 2A). In contrast, when we crossed the AAEL010097-Cas9 female and U6-sgRNA male, the larvae that developed showed the mosaic eye phenotype (Fig. 2A). In two independent experiments, 73% (43/59) and 82% (82/100) of insects showed the mosaic eye phenotype.
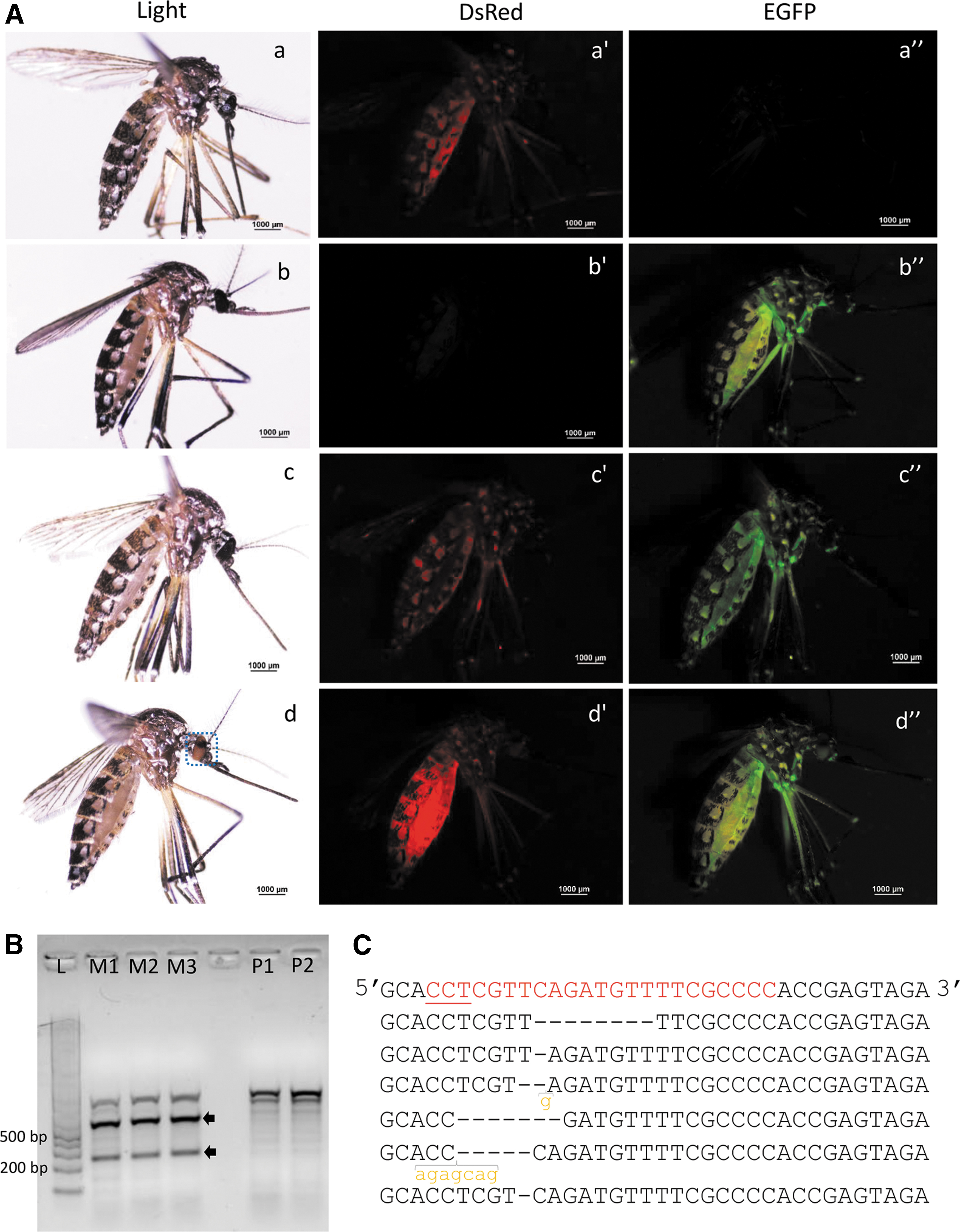
Maternally inherited Cas9 induced mutations in the kmo gene of Aedes aegypti.
To confirm mutations in the kmo gene, the genomic DNA from the progeny of both crosses was extracted, and the T7E1 assay was performed. As shown in Figure 2B, progeny from the Cas9 female and U6-kmo-sgRNA male cross showed mutations in the kmo gene. In contrast, the progeny from the Cas9 male and U6-kmo-sgRNA female cross did not show any mutations in kmo gene. The PCR fragments were cloned and sequenced. Six out of the eight clones sequenced showed mutations, including 1, 7, or 8 bp deletions and two different indels (Fig. 2C). Notably, mutations were only induced in the offspring when the Cas9 line female was mated with the U6-kmo-sgRNA male. The AAEL010097 promoter used to drive the expression of Cas9 is predominantly expressed in the ovary and early embryonic development, 22 which may have contributed to the requirement of maternal Cas9 for successful mutagenesis. 26 However, non-Mendelian dominant maternal effects caused by CRISPR-Cas9 transgenic expression was reported in Drosophila melanogaster. 27 The dominant maternal effects induced by CRISPR-Cas9 warrants further investigation.
Target-specific mutagenesis of Met using the transgenic CRISPR-Cas9 system
The design of sgRNA targeting exon 4 of the juvenile hormone receptor, Methoprene tolerant (Met), was reported previously (Fig. 3A). 18 The U6-Met-sgRNA transformation vector was constructed as described above for U6-kmo-sgRNA (Fig. 3B). The U6-Met-sgRNA line was generated by injecting U6-Met-sgRNA plasmid and transposase mRNA into 400 Liverpool IB12 embryos. The transgenic insects were selected, and the expression of sgRNA was checked by reverse transcription qualitative PCR using Met-specific primers. The expression of Met-sgRNA was detected in U6-Met-sgRNA transgenic insects (Supplementary Fig. S1).
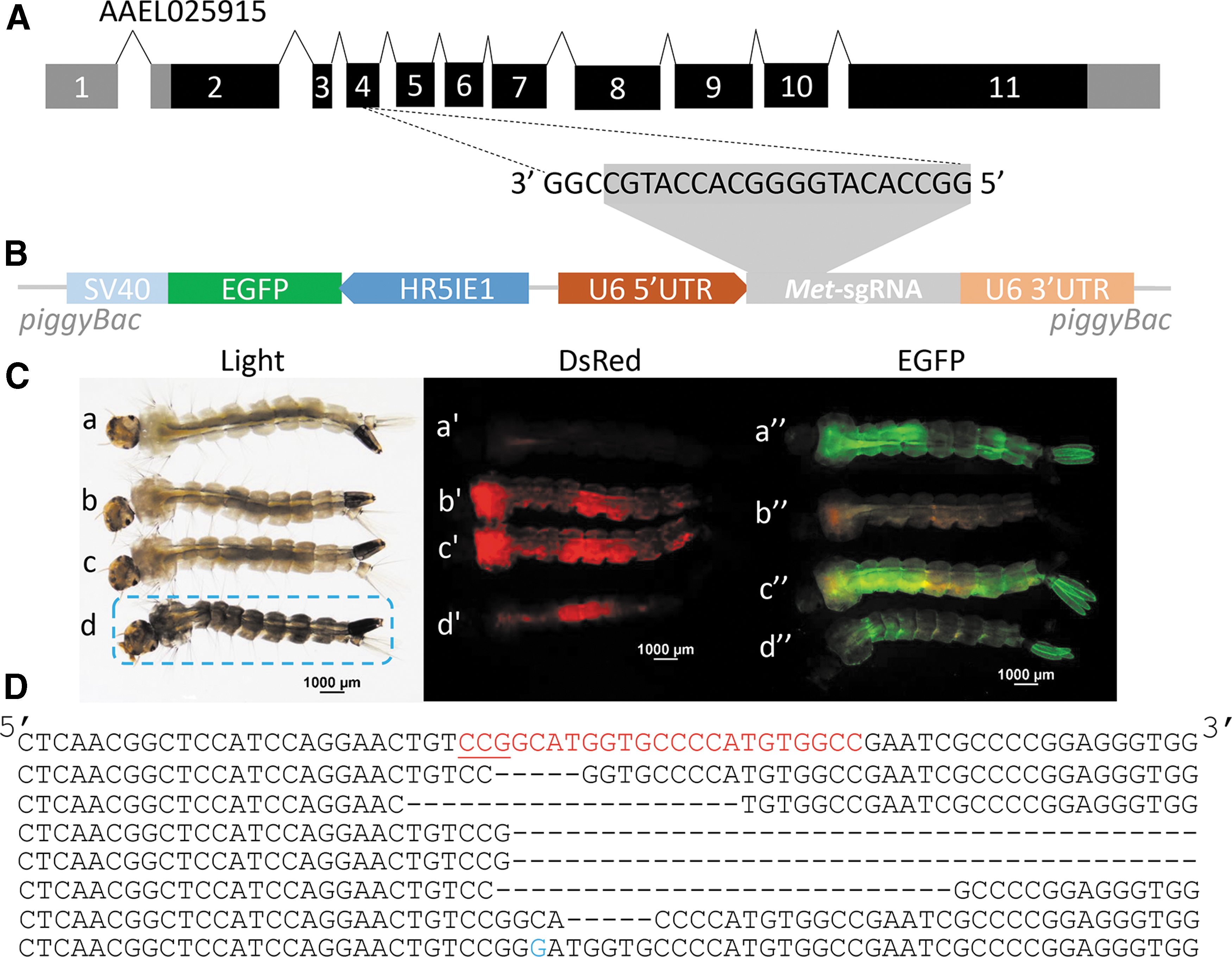
The dualistic Cas9/sgRNA system induced mutations in the Met gene of Ae. aegypti.
The U6-Met-sgRNA line was mated with the AAEL010097-Cas9 line, and the progeny were evaluated for the Met mutant phenotype. As shown in Figure 3C, the black larval phenotype of Met knockout 18 was detected only in the larvae developed from Cas9 female and U6-Met-sgRNA male crosses. Similar to the results from kmo mutagenesis, only the maternally inherited Cas9 induced mutations in the Met gene. In two independent experiments, 94.4% (34/36) and 63.6% (14/22) of individuals displayed a black larval phenotype during the third and fourth later larval stages. The mutations of the Met gene caused by the transgenic system were confirmed by TA cloning and sequencing. Eight clones sequenced showed six different deletions and one transmission (Fig. 3D).
In a previous study, we used the multiple sgRNAs method to knock out the Met gene in one generation. 18 To increase the mutation frequency, one of the kmo sgRNA was mixed with the multiple Met sgRNAs to identify the mutants before they showed the black larval phenotype. With the method we used here, using the DsRed and eGFP fluorescent marker, we could identify the Met mutants before the onset of the black larval phenotype using the DsRed and eGFP signals. Similar approaches could be used to identify mutants of target genes without pronounced phenotype.
CRISPR-Cas9 genome editing systems have been developed for various applications in mosquitoes.8,12,22,23,28–36 Transgenic approaches for expression of Cas9 and sgRNA have been developed for use in D. melanogaster 37 and Bombyx mori. 28 The recent studies on Ae. aegypti employed transgenic expression of sgRNA and Cas9 for the development of gene-drive systems.23,38 Here, we used the transgenic expression of sgRNA and Cas9 for improving this system for functional genomics studies in Ae. aegypti. The multiple sgRNA combined with a marker sgRNA method was described previously, 18 and the transgenic CRISPR-Cas9 method described here adds to the CRISPR-Cas9 toolbox of Ae. aegypti and other insects, and will facilitate functional genomics studies in mosquitoes and other insects.
Footnotes
Acknowledgments
We thank Jeff Howell for help with insect rearing and reading the earlier version of the manuscript, and Robert A. Harrell II, Channa Aluvihare, and Omar S. Akbari for help with embryonic injections, Ae. aegypti rearing, and the AAEL010097-Cas9 strain.
Author Disclosure Statement
The authors declare that no competing financial interests exist for all authors of this manuscript.
Funding Statement
This work was supported by grants from the National Institutes of Health (GM070559-14) and the National Institute of Food and Agriculture, U.S. Department of Agriculture (under HATCH Project 2351177000).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.