
Abstract
Allele-specific genomic targeting by CRISPR is a versatile strategy that has been increasingly exploited not only in treating inherited dominant diseases and mutation-driven cancers, but also in other important fields such as genome imprinting, haploinsufficiency, and genome loci imaging. Despite its tremendous utilities, few bioinformatic tools have been implemented for the allele-specific purpose of CRISPR. We thus developed AsCRISPR (Allele-specific CRISPR), a comprehensive web tool to aid the design of short-guide RNA (sgRNA) sequences that can discriminate between alleles. AsCRISPR allows users to analyze both their own identified variants and heterozygous single nucleotide polymorphisms and, importantly, output the candidate sgRNAs and their quality control information. To facilitate targeting dominant diseases, AsCRISPR analyzed dominant single nucleotide variants (SNVs) retrieved from ClinVar and OMIM databases, and generated a dominant database of candidate-discriminating sgRNAs that may specifically target the alternative allele for each dominant SNV site. Moreover, a validated database was established, which manually curated the discriminating sgRNAs that were experimentally validated in the mounting literature for multiple allele-specific purposes.
Introduction
Inherited diseases are caused by various types of mutations, insertions/deletions (indels), large genomic structural variations, as well as pathogenic single nucleotide polymorphisms (SNPs) that are critical for precision medicine. Of those, dominantly inherited diseases present a special challenge for researchers to conduct gene therapies. Such patients inherited only one mutated allele and one normal allele on pairs of chromosomes. The treatment strategy typically involves an allele-specific manipulation by silencing or ablating the pathogenic alleles while exerting no aberrant effects on the wild-type ones. Previously, mounting studies have used allele-specific short interference RNAs tosuppress dominant mutant alleles selectively, with promising therapeutic benefits.1,2 Until recent years, allele-specific CRISPR genome editing has emerged as a promising means to treat human genetic disorders and cancers, and has also been increasingly implemented in other versatile applications such as genome imprinting, haploinsufficiency, genome loci imaging, and immunocompatible manipulations. 3 The CRISPR system provides highly specific genome editing that is capable of discriminating disease-causing alleles from wild-type ones, whenever the genetic variants are (1) in PAM and generate unique protospacer adjacent motifs (PAMs) or (2) near PAM, located within the spacer region, especially the seed region of short-guide RNAs (sgRNAs).3,4
So far, allele-specific CRISPR has been increasingly employed in treating various diseases such as retinitis pigmentosa,5–8 corneal dystrophy, 9 dominant progressive hearing loss, 10 and multiple mutation-driven cancers,11–13 as well as genome imprinting diseases. 14 It has also been used to alleviate haploinsufficiency by allele-specific CRISPR activation of wild-type alleles, 15 and has even been designed for manipulating human leukocyte antigen locus. 16 More excitingly, this strategy has recently been utilized to inactivate mutant huntingtin (mHtt) selectively, taking advantage of novel PAMs created by SNPs flanking the HTT locus.17,18 Overall, allele-specific CRISPR is now believed to be a promising personalized strategy for treating genetic diseases.
However, it is always labor intensive and time-consuming to figure out appropriate sgRNAs that may discriminate between two alleles.9,17 Currently, most web servers only design sgRNAs from the reference genomes, without allele discriminations. Thus, we developed AsCRISPR (Allele-specific CRISPR), a web server to aid the design of sgRNAs for allele-specific genome engineering. AsCRISPR is freely available at http://www.genemed.tech/ascrispr. It has incorporated multiple CRISPR nucleases and can process flexibly with either user-identified variants or heterozygous SNPs. AsCRISPR facilitates the selection of optimal discriminating sgRNAs, with the output of multiple on-target scores and elaborated off-target candidates. Importantly, to facilitate targeting dominant diseases, for the first time, we analyzed with dominant single nucleotide variants (SNVs) retrieved from the ClinVar and OMIM databases, and generated a dominant database of candidate-discriminating sgRNAs that may specifically target the alternative alleles for each dominant SNV site. Meanwhile, we are in the ongoing process of collecting experimentally validated allele-specific sgRNAs reported in the mounting literature for disease treatments and many other applications.
Methods
AsCRISPR was developed using PHP and Perl on a Linux platform with an Apache web server. The front and back separation model was used; the front end is based on the Vue + Element, and the back end is based on the Laravel, a PHP web framework.
Single-base mutations, short indels, and SNP IDs are the formats for input (Fig. 1). The SNP information was downloaded from the dbSNP v150 database (https://www.ncbi.nlm.nih.gov/SNP) and stored in MySQL database. To optimize the SNP query performance, an index on SNP table was added. The sequence can be extracted from the .2bit file (hg19/GRCh37, hg38/GRCh38, or mm10/GRCm38) with the twoBitToFa command base on the SNP information (chromosome, start genomic position and end genomic position, reference allele, and alternate allele). AsCRISPR displayed the SNP sites located at both flanking nucleotides of a query SNP ID, which was implemented using D3.
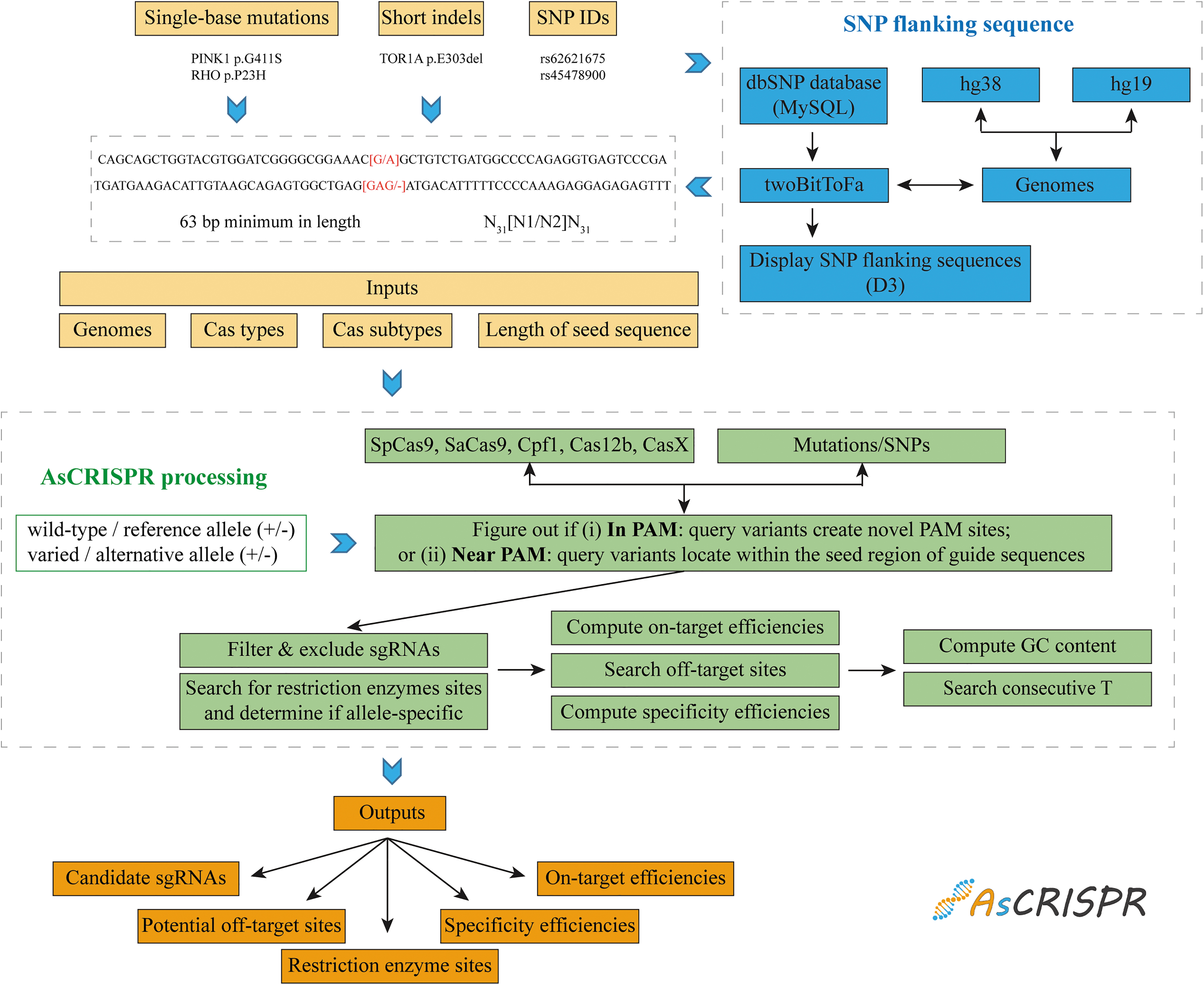
Workflow of AsCRISPR. Color images are available online.
In principle, AsCRISPR proceeds to figure out if (1) in PAM query variants give rise to novel PAMs, which confers stringent allele-specific targeting, or (2) near PAM query variants are located within the seed region of sgRNAs, which may abolish the Cas cleavage (Fig. 1 and Supplementary Fig. S1A). AsCRISPR then outputs the candidate sgRNAs, after performing the stringent search and filter. For example, those sgRNAs with novel PAMs generated by variants that constitute an ambiguous genetic code (such as R and Y in the CjCas9:NNNNRYAC) will be excluded.
Scripts from CRISPOR (https://github.com/maximilianh/crisporPaper) were then integrated into AsCRISPR to assess sgRNA properties and scores. AsCRISPR also searched for possible sites recognized by restriction enzymes deposited in our database. In addition, sgRNAs were further analyzed and cautioned as “not recommended” if (1) the GC contents are beyond 20–80% or (2) contain four or more consecutive T, which might terminate the U6 or U3 promoter-driven transcription.
The data set for the dominant database was sourced from the ClinVar database. 19 We chose “pathogenic” and “pathogenic/likely pathogenic” variants deposited in the ClinVar database after June 6, 2020. Autosomal dominant information of variants was extracted from Online Mendelian Inheritance in Man (OMIM, V20190524). 20
The source codes of AsCRISPR are available on Github: https://github.com/zhaoguihu/ascrispr
Results
AsCRISPR helps to design sgRNAs based on four major types of Cas nucleases, including the commonly used Cas9, Cpf1, and also recently reported Cas12b21,22 and CasX, 23 each type of which contains its variant subtypes with distinct PAM sites and seed lengths (Table 1). This allows the users to choose the optimal combination of Cas protein and sgRNA freely to meet their own needs. However, some extra caution might be noticed that studies also documented cases of CRISPR editing at non-canonical PAMs, such as NAG for SpCas9, albeit with much lower efficiency.24,25
PAM, protospacer adjacent motif.
Input format
The inputs for AsCRISPR could be DNA sequences harboring single-base mutations or short indels, and also simply SNP IDs deposited in the dbSNP database. All inputs will be finally processed as the format of N31[N1/N2]N31, in which N1/N2 denotes the sequence in the wild-type/reference and mutated/alternative allele, respectively. Thus, it means that the input sequence requires a minimum length of 63 bp, with at least 31 bp flanking the variant site, to be processed for the output of a complete list of candidate discriminating sgRNAs (Supplementary Fig. S1B and C). Notably, when users query an SNP ID, AsCRISPR will also display other SNP sites located at both flanking 31 nucleotides (Supplementary Fig. S1D), which provides extra variation information and would be of great value for designing personalized genome targeting.
Candidate sgRNAs
AsCRISPR provides downloadable results with candidate sgRNAs that target only one allele (Fig. 2). For better visualization, AsCRISPR ranks all sgRNAs first by listing pairs with the same PAM sequence back to back. Furthermore, AsCRISPR evaluates their on-target efficiencies, specificity efficiencies, and potential off-targets throughout the genome, taking advantage of CRISPOR's scoring system. 26 Specifically, on-target efficiencies were calculated with multiple reported algorithms and were normalized to 0–1. For SpCas9, efficiency scores were predicted according to Xu et al., 27 Doench et al., 28 Moreno-Mateos et al., 29 and Listgarden et al. 30 For SaCas9, efficiency scores were predicted according to Najm et al. 31 For Cpf1, Cas12b, and CasX, efficiency scores were predicted according to Kim et al. 32
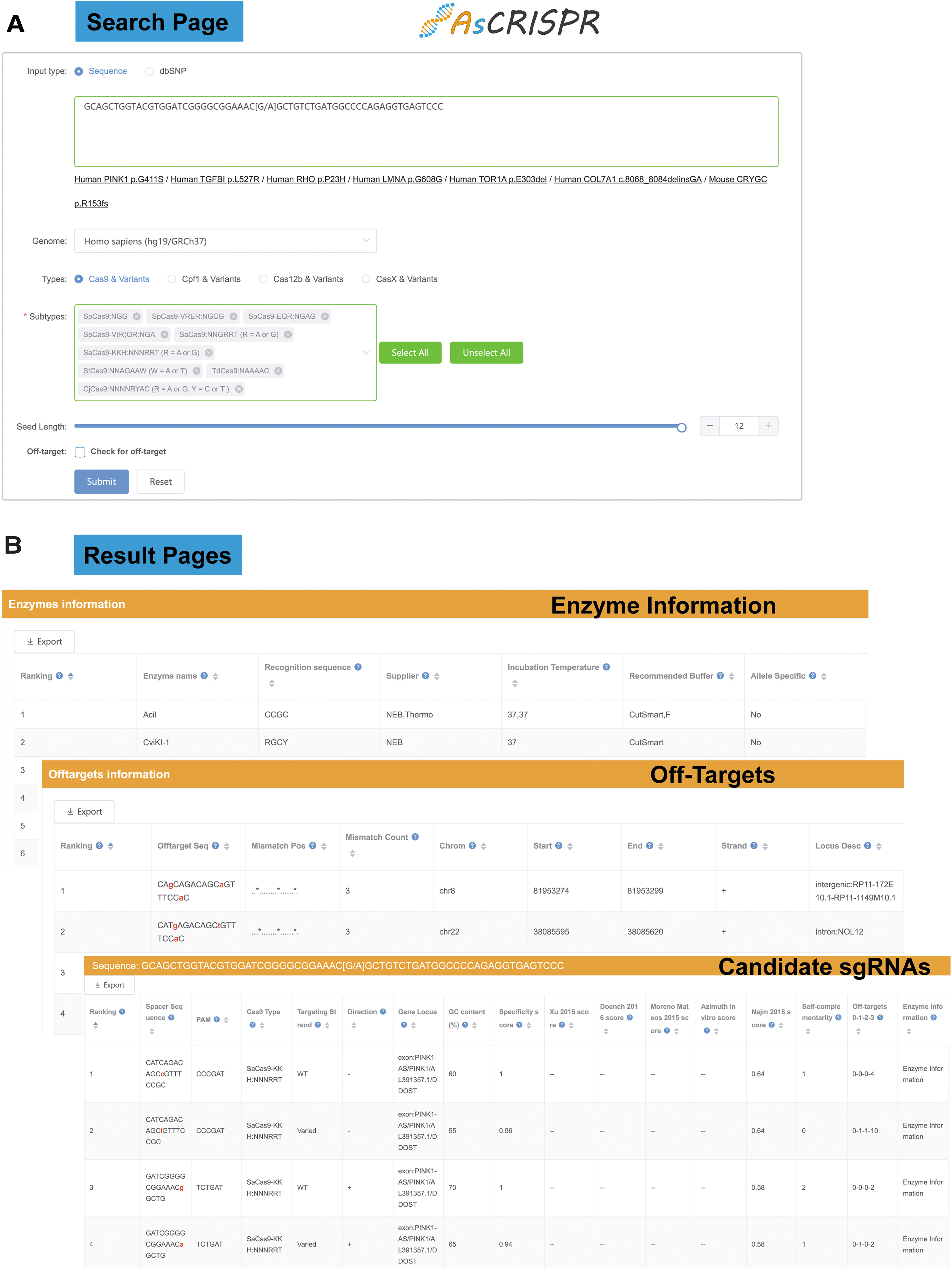
Snapshot of the graphic user interface of AsCRISPR.
Off-target sequences
The potential off-target sequences throughout the genome are rigorously searched by a maximum of three-base mismatches (Fig. 2). AsCRISPR lists the number of off-target sequences for each sgRNA with 0, 1, 2, or 3 mismatches (0-1-2-3). Clicking on the 0-1-2-3 will reveal more information about the off-target information in the downstream data sheet, including the locations (exon, intron, or intergenic region), sequence mismatches, and so forth. Users can freely re-rank the off-target sequences by locations. The specificity score measures the uniqueness of a sgRNA in the genome. The higher the specificity score, the lower are off-target effects. Specificity scores were calculated based on Hsu scores 24 and CFD scores. 28 For Cpf1, Cas12b, and CasX, no off-target ranking algorithms were available in the literature so far. Instead, we just applied Hsu and CFD scores to their off-target sequences.
Restriction sites
AsCRISPR also searches for possible sites recognized by restriction enzymes, along with the spacer sequences (Fig. 2), which might be disrupted after gene targeting, and further determines whether those candidate enzymes are also allele specific. This provides an important tool for the characterization and screening of targeted single colonies by restriction fragment length polymorphism (RFLP).
Exemplary running
We have listed several typical sequences on the Web site for exemplary running. For example, heterozygous PINK1 p.G411S is one of the ideal mutations for allele-specific targeting, which was previously demonstrated to increase the risk of Parkinson's disease via a dominant-negative mechanism. 33 In the Cas9 mode, AsCRISPR outputs 11 discriminating sgRNAs in combination with three subtypes of Cas9, including SpCas9, SpCas9-V(R)QR, and SaCas9-KKH (Supplementary Table S1A). One of those sgRNA exploits a novel PAM (5′-CgG-3′) created by the mutation, and another five pairs of sgRNAs containing the mutation point within the seed region selectively target either wild-type allele or mutated allele (Supplementary Table S1A). Therefore, by using Cas9, a total of five candidate sgRNAs might be specific to the mutated PINK1 p.G411S allele, which is ready for the users' experimental evaluations (Supplementary Table S1A). Besides, we have also listed other exemplary mutations, including the single mutations (TGFBI p.L527R; RHO p.P23H; LMNA p.G608G), three-base delete mutations (TOR1A p.E303del), and short indel mutations (COL7A1 c.8068_8084delinsGA).
Similarly, for heterozygous SNPs, AsCRISPR processes the input SNP numbers and translates them into DNA sequences (63 bp) after retrieving the genomic database. As an example, we used AsCRISPR to analyze one of the SNPs, rs63750526:[C>A], only with Cas9, and successfully obtained 15 discriminating sgRNAs in combination with seven subtypes of Cas9: SpCas9, SpCas9-V(R)QR, SpCas9-EQR, SpCas9-VRER, SaCas9, SaCas9-KKH, and St3Cas9 (Supplementary Table S1B). Five of those sgRNA exploit novel PAMs (5′-TGcG-3′, 5′-TGaG-3′, 5′-TGa-3′; 5′-CTGaGT-3′) created by the SNP, and the remaining sgRNAs contain the variant point within their seed sequences (Supplementary Table S1B).
Users can freely re-rank the candidate sgRNAs by Cas types, on-target efficiencies, specificity scores, off-target properties, and others. We recommend selecting the sgRNAs with novel PAMs, since they contribute to the most stringent discrimination. For a more detailed demonstration, users can also find an AsCRISPR tutorial on the Web site, which can be read online or downloaded as a PDF document.
Databases
To strengthen the purpose of targeting dominant diseases, AsCRISPR collected dominant SNV sites from ClinVar and OMIM databases for allele-specific analysis, and for the first time generated a dominant database depositing candidate targetable sgRNAs, which may specifically target the alternative allele for each dominant SNV site. Notably, we observed that the ClinVar and OMIM databases are not correctly annotated, in several cases, with the dominant nature of diseases. Thus, several clearly dominant diseases/SNV sites might be filtered out from the dominant database. Nevertheless, in the current version of AsCRISPR, we collected 102,212 records of dominant SNV sites (with 12,938 SNV sites pathogenic or likely pathogenic), related to 1,833 different diseases. AsCRISPR analyzed the dominant SNV sites and found that around 36.96% (38.25% for pathogenic dominant SNVs) are targetable by allele-specific CRISPR (Fig. 3A). Among the four major types of Cas nucleases, Cas9 is the most frequently employed, probably due to its multiple subtypes (Fig. 3B). Further analysis showed that four Cas9 subtypes (SpCas9, SpCas9-V(R)QR, SaCas9-KKH, and Nme2Cas9) are obviously more robust in targeting SNV sites, which is consistent with the simpler nature of PAM sequences (Fig. 3C). Generally, the alternative allele of each dominant SNV site can be targeted by an average of 14.15 discriminating sgRNAs. Among those, 2.84 sgRNAs are in PAM (create a novel PAM site) and 11.57 are near PAM (in the spacer seed region; Fig. 3D). Interestingly, the major types of Cas nucleases that contribute to the stringent in PAM targeting are SpCas9, SpCas9-V(R)QR, SaCas9-KKH, and Nme2Cas9 (Fig. 3E). Basically, the targeting features are similar between dominant SNVs and pathogenic dominant SNVs (Fig. 3).
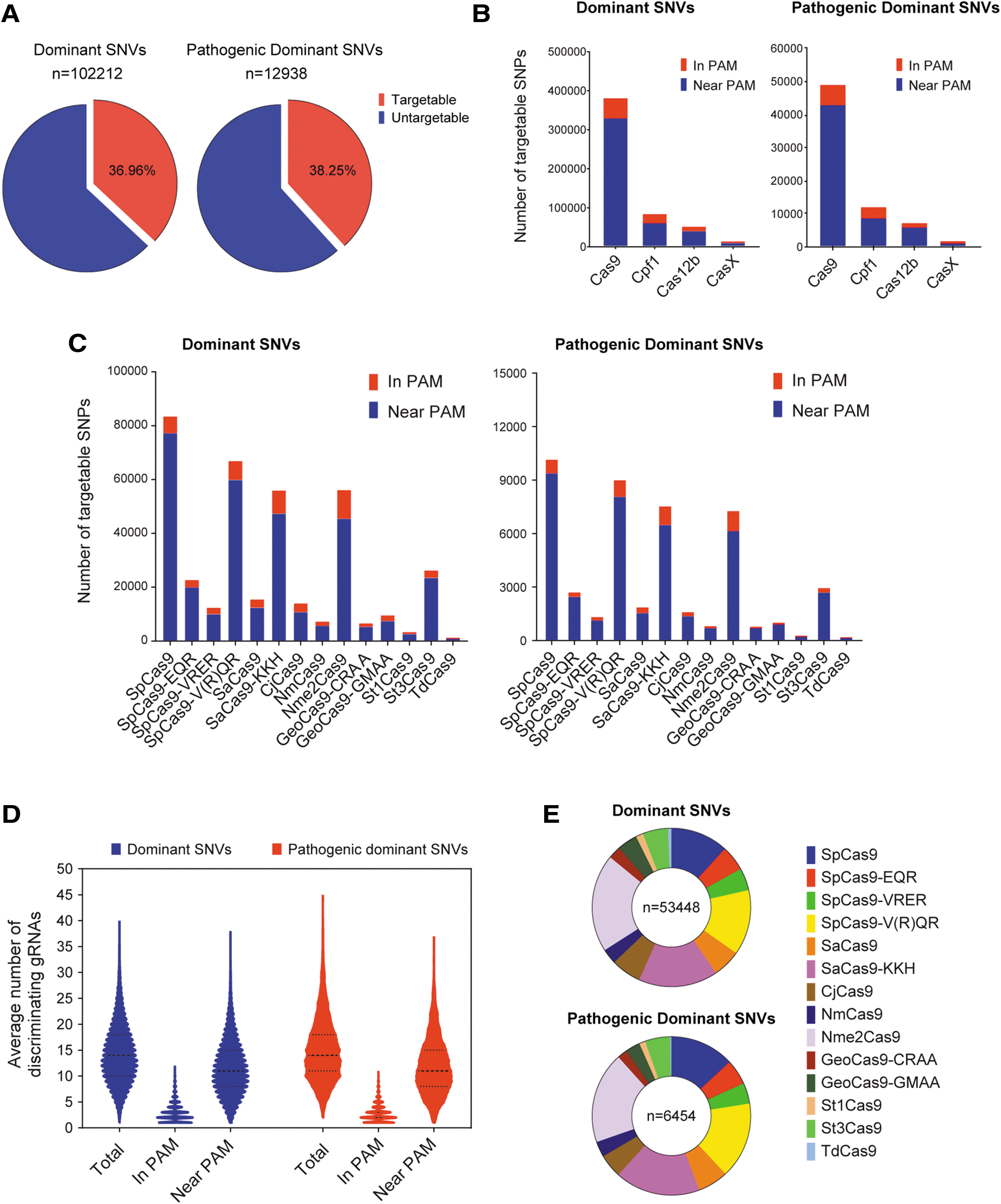
Statistics of dominant database.
Considering the predictive nature of the dominant database, we also manually curated the experimentally validated sgRNAs that were reported in the mounting literature for disease treatments and other allele-specific purposes, and thus established a validated database. This database is expected to be expanded rapidly along with the advancements in this field.
Future Developments
So far, AsCRISPR has integrated the genomes of Homo sapiens (hg19/GRCh37), Homo sapiens (hg38/GRCh38), and Mus musculus (mm10/GRCm38). We are planning to upload more genomes for analysis in the near future in order to expand its allele-specific utilities. However, users can still input their identified sequences or DNA sequences from other species. The current version of AsCRISPR would then output the discriminating sgRNAs, but without displaying the information of gene locus, scores, and off-target sequences.
Increasing the types of Cas nucleases such as ScCas9, 34 xCas, 35 SauriCas9, 36 and recently developed Cas12j, 37 as well as those targeting on RNAs such as CasRx 38 and Cas13a39,40 (already reported with allele-specific purpose), will be added for expanded utilities. Moreover, AsCRISPR is going to be exploited for increasing allele-specific applications, for example by combining with base editors,41–44 transposases,45,46 and the emerging prime editors. 47 For instance, base editors have been recently used to introduce stop codons that will lead to early translation termination,48,49 or to change the alternative mRNA splicing by targeting the exon–intron boundary.50,51 It is also hopeful that those newly developed applications will be implemented in an allele-specific manner.
Our understanding of on- and off-target sgRNA efficiencies is evolving rapidly. Although the on-target efficiencies in AsCRISPR were calculated with multiple reported algorithms, the scoring algorithms have been continuously improved. Cas12b and CasX may have special efficiency scoring algorithms that are different from those of Cas9 and Cpf1. However, to the best of our knowledge, there is still a lack of published studies on this. We will thus incorporate the convincing scoring algorithms, which predict either on- or off-target efficiencies, into AsCRISPR once they become available.
AsCRISPR will be updated in a timely manner, along with the emerging exploitation of the Cas toolbox. Moreover, the dominant database and validated database resources will be updated three times per year. We also welcome users to send us their experimentally validated discriminating sgRNAs for our update. All in all, we welcome any constructive feedback from users for improving our web server.
Discussion
Comparison with similar servers
We have thus developed AsCRISPR, which is an easy-to-use and streamlined web tool for designing potential discriminating sgRNAs between alleles to facilitate CRISPR-based personalized therapy and other versatile allele-specific applications. In particular, we have incorporated two recently reported types of Cas nucleases, Cas12b and CasX, which have been shown to be promising for genetic engineering due to their smaller size and higher specificity. Just as we just finished the AsCRISPR implementation, another software termed AlleleAnalyzer was published, aiming to identify optimized personalized and allele-specific sgRNAs. 52 AlleleAnalyzer also leverages patterns of shared genetic variation across thousands of publicly available genomes to design sgRNA pairs that will have the greatest utility in large populations. 52 However, the difference is that as a web tool, AsCRISPR can process either user-identified sequences or SNP numbers, which is more likely to be demand driven for research studies and clinical therapeutics. Moreover, AsCRISPR only outputs single sgRNAs instead of pairs of sgRNAs, although users may also freely use AsCRISPR to design another sgRNA manually to make a sgRNA pair. Note that as numerous non-coding RNAs or regulatory elements are widespread, dual-sgRNA excision of a large DNA fragment might bring about extra risks for disease treatment. Thus, AsCRISPR may possess extra allele-specific utilities and add to the bioinformatic repositories for allele-specific genomic editing.
Another two web servers, SNP-CRISPR and CrisPam, have also been developed recently, converging with our web tool, AsCRISPR, but at different publication status, to share the same notion of the allele-specific designing of sgRNAs. Basically, SNP-CRISPR designs sgRNAs based on the public SNP database yet with only two types of PAM. 53 The other server, CrisPam, only analyzes the variants that produce novel PAMs (in PAM) from pathogenic and likely pathogenic SNPs in humans. 54 Interestingly, they found that 84% of the total SNPs checked can create novel PAMs, and the average number of PAMs generated by an SNP is 6.97. 54 In contrast, analysis of the results with our database show that an average of 2.84 sgRNAs are in PAM. There are a number of reasons for this difference. We analyzed only dominant SNVs from ClinVar and OMIM databases, which underlies any potential differences. Yet, the major difference is the type of Cas proteins used for analysis. In CrisPam, multiple Cas with simpler PAMs such as xCas were involved. Regarding this, the results from our database are still underestimated. Nevertheless, more tool Cas proteins with expanded PAM sequences are needed in order to achieve the full potential of allele-specific genome targeting.
The brief properties of different servers were summarized in Table 2. AsCRISPR aims to be the most comprehensive web tool for allele-specific purposes, endowed with most Cas nucleases and PAM sites, selectable seed lengths, importantly quality control information of candidate sgRNAs such as multiple on-target scores, elaborated off-target sequences and RFLP sites, as well as curated databases of targetable dominant SNVs and experimentally validated discriminating sgRNAs. However, the current version of AsCRISPR cannot process with a batch query due to the limited configuration of our platform server, and the response time is slower than it is for SNP-CRISPR and CrisPam, given that AsCRISPR searches genome-wide off-target sequences for a set of candidate sgRNAs. Those disadvantages are going to be addressed in the future, as we are continuously upgrading our platform server.
Also has a command-line version.
SNP, single nucleotide polymorphism; sgRNA, single guide RNA; WT, wild type.
Population versus personalized therapy
Interestingly, people have been avoiding genetic variants when designing sgRNAs for therapeutic genome editing in large populations for a long time. Previous studies performed a comprehensive analysis of the Exome Aggregation Consortium (ExAC) and 1000 Genomes Project (1000GP) data sets, and determined that genetic variants could negatively impact sgRNA efficiency, as well as both on- and off-target specificity at therapeutically implicated loci.55,56 Thus, for CRISPR-based therapy in large patient populations, genetic variations should be considered in the design and evaluation of sgRNAs to minimize the risk of treatment failure and/or adverse outcomes. To address that, people thus endeavor to identify universal/platinum sgRNAs located in the low-variation regions, with the help of, for example, the ExAC or gnomAD browser, to maximize their population efficacy.55,57 Fortunately, several tools have been available that take into account genetic variations such as WGE 58 or CRISPOR 26 during the sgRNA design.
Although the genetic variations would be a challenge for platinum sgRNA design, it provides a promising entry for designing allele-specific or personalized sgRNAs in treating individual patients. Deciphering genetic variations helps to seek common platinum sgRNAs for treatment in large populations, whereas tools including AsCRISPR have the opposite aim that exploits the discrimination abilities of heterozygous genetic variants to facilitate the design of allele-specific targets for individuals.
The rapid advancements of genome sequencing technologies have accelerated the research of variants and their physiological functions or effects in the era of precision medicine. Specifically, with the expanding toolkits of the CRISPR system, emerging studies have been performed regarding the variants linking with the pathogenesis of human diseases. Apart from treating genetic diseases, allele-specific CRISPR genome editing has also been increasingly used in other research areas such as genome imprinting, haploinsufficiency, spatiotemporal loci imaging, and immunocompatible manipulations, and, for sure, will be exploited for more applications in the near future. Therefore, AsCRISPR will be right on track, along with the rapidly progressing research on genetic variants.
Footnotes
Acknowledgments
An earlier draft of this manuscript was posted at bioRxiv (DOI: 10.1101/672634).
Author Disclosure Statement
The authors declared no competing interests.
Funding Information
This work was supported by grants from the National Natural Sciences Foundation of China (81801200 to Y.T.); talents startup funds of Xiangya Hospital (2209090550057 to Y.T.); and Hunan Provincial Natural Science Foundation of China (2019JJ40476 to Y.T., 2019JJ50974 to G.Z.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.