
Abstract
Among current reported Cas12a orthologs, Francisella novicida Cas12a (FnCas12a) is less restricted by protospacer adjacent motif (PAM). However, the activity of FnCas12a nuclease is relatively low or undetectable in human cells, limiting its application as desirable genome engineering tools. Here, we describe TEXT (Tethering EXonuclease T5 with FnCas12a)—a fusion strategy that significantly increased the knockout efficiency of FnCas12a in human cells at multiple genomic loci in three different cell lines. TEXT results in higher insertion and deletion efficiency than FnCas12a under different spacer lengths from 18 nt to 23 nt. Deep sequencing shows that TEXT substantially increased the deletion frequency and deletion size at the targeted locus. Compared to other Cas12a orthologs, including AsCas12a and LbCas12a, TEXT achieves the highest on-targeting efficiency and shows minimal off-targeting effects at all tested sites. TEXT enhances the activity of FnCas12a nuclease and expands its targeting scope and efficiency in human cell genome engineering.
Introduction
CRISPR-Cas adaptive immune systems protect bacteria or archaea from viral invasion. 1 Leveraging CRISPR-Cas systems, a variety of genome engineering technologies have been developed, greatly accelerating the study of synthetic biology, gene therapy, diagnostics, plant engineering, and so on.2–6 The development of alternative CRISPR nucleases other than commonly used Streptococcus pyogenes Cas9 (spCas9) would expand the genome engineering toolbox with new and potentially advantageous properties.7–9 The CRISPR-Cas12a system has been developed for genome editing applications with distinct features, such as recognizing T-rich protospacer adjacent motif (PAM) sequences, cleaving DNA with staggered cut distal to 5′ T-rich PAM, and the ability to process its own crRNA arrays by Cas12a.9–12 In addition, in contrast to Cas9 that uses crRNA/tracrRNA hybrid for DNA targeting, Cas12a only requires a single short (about 40 nt) CRISPR RNA (crRNA), which is easier to prepare. 9 Cas12a orthologs from Acidaminococcus sp. BV3L6 (As), Lachnospiraceae bacterium (Lb) have been used for genome engineering in many organisms, including human cells.9–13 However, the targeting range of AsCas12a and LbCas12a has been hindered by the strict requirement of a TTTV PAM sequence. Although several protein engineering strategies have generated new AsCas12a variants that can target non-canonical PAMs, including TATV, TYCV, TTYN, VTTV, and TRTV, there are still a large number of genomic sites that cannot be accessed by those variants.14,15 FnCas12a has been reported to use shorter and more frequently occurring PAM sequences (TTN or TTV) in vitro and in human cells,9,16,17 which is less restricted than AsCas12a and LbCas12a, and holds promise for targeting more currently inaccessible genomic regions. However, the nuclease activity of FnCas12a is relatively lower or unobservable in human cells.16,17 Therefore, strategies that can improve the editing efficiency of FnCas12a will give full play to its advantages of wide targeting ability.
To date, two types of engineering strategies have been used to increase the knockout efficiency of CRISPR-Cas12a, either enhancing the nuclease activity of Cas12a by rational protein mutagenesis or improving the stability of crRNA by optimizing its secondary structures.18–21 However, neither of those approaches can fundamentally leverage the decisive step of gene knockout, the endogenous non-homologous end joining (NHEJ) DNA repair pathway. Once the DNA double-strand break (DSB) is created at the target locus, the NHEJ pathway will be responsible for ligating the break ends back together. Typically, NHEJ will use short homologous DNA sequences referred to as microhomologies to guide the repair. The microhomologies usually exist in the form of single-strand overhangs at the DSB ends. 22 When the overhangs are perfectly compatible, NHEJ usually repairs the break accurately, without causing insertions and deletions (indels).23–25 However, the imprecise NHEJ repair, which contributes to the knockout efficiency, can disrupt genes by randomly introducing unwanted indels at the ligation joint, occurring much more commonly when the overhangs are not compatible.22,23,26–28 FnCas12a has been characterized to generate staggered, perfectly compatible 5′ single-strand overhangs at DSB ends, 9 which is a desired substrate for error-free rather than indel-involved NHEJ repair. Therefore, we reason that by resecting the 5′ single-strand overhang at the DSB ends, the NEHJ pathway can be biased to use the error-prone repair more frequently, thereby improving the overall knockout efficiency of FnCas12a.
Here, to improve the editing efficiency of FnCas12a in human cells, we fused T5 ssDNA exonuclease to N terminal of FnCas12a, namely TEXT (Tethering EXonuclease T5 with FnCas12a), which can direct the cellular NHEJ repair more frequently into the error-prone pathway, enabling higher indel efficiency than when using FnCas12a alone. TEXT exhibits two- to threefold higher genome editing activity than FnCas12a at all tested sites, and also increases the deletion size under different spacer lengths from 18 to 23 nt, in which 18 nt results in the highest fold increase. Notably, when using 18 nt spacer crRNA, the deletion size of TEXT is mainly 8 or 9 nt. In addition, compared to FnCas12a, LbCas12a, and AsCas12a, TEXT showed the highest editing efficiency among all test sites with minimal off-target effects. In conclusion, we developed an improved FnCas12a system that has a wide range of genome editing applications, which will help target previously inaccessible genomic sites.
Methods
Plasmid construction
Polymerase chain reaction (PCR) was performed using Q5 high-fidelity DNA polymerase (New England Biolabs, Ipswich, MA), and specific primers were synthesized by Genewiz (Suzhou, PR China). Human codon-optimized exonuclease from phage T5, phage T7, and phage λ were synthesized by Genewiz. The Artemis gene was amplified from complementary DNA (cDNA) of HEK293T cells. Recj and PolA-exo were amplified from Escherichia coli genomic DNA. Briefly, the FnCas12a/EXO-FnCas12a driven by the cytomegalovirus promoter and crRNA expression cassette driven by the U6 promoter were constructed into one plasmid base on the backbone of pcDNA3.1(+) plasmid and separated by a spacer sequence. The amino acid sequence of TEXT is shown in Supplementary File S1, and the sequences of crRNA used in this study are listed in Supplementary File S2.
Human cell culture and transfection
HEK293T cells and Hela cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin and streptomycin (Solarbio, Beijing, PR China). HLEB3 cells were cultured in DMEM supplemented with 15% FBS and 1% penicillin and streptomycin. Cells were maintained at 37°C with 5% CO2. Cells were seeded onto 96-well plates (Corning, Corning, NY) and transfected at approximately 60% confluency. Plasmids (200 ng) were transfected using Lipofectamine 2000 (Life Technologies, Carlsbad, CA) following the manufacturer's recommended protocol.
Genomic DNA preparation and PCR amplification
Sixty hours after treatment, cells were washed with PBS genomic DNA extraction. Genomic DNA was extracted using the QuickExtract DNA Extraction Solution (Epicentre Technologies, Madison, WI) following the manufacturer's protocol. Concentrations of gDNA were determined on a Nanodrop 2000. Genomic regions flanking the target sites were amplified using 200 ng purified gDNA template, Q5 high-fidelity DNA polymerase, and specific primers (Supplementary File S2 and S4) on a T100 thermal cycler (Bio-Rad, Hercules, CA).
T7E1 cleavage assay
PCR fragments were generated using Q5 high-fidelity DNA polymerase, and then hybridized in NEB Buffer 2 (50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM DTT, pH 7.9; New England Biolabs) by heating to 98°C for 3 min, followed by a 2°C/s ramp down to 85°C, 1 min at 85°C, and a 0.1°C/s ramp down to 25°C on a T100 thermal cycler. Subsequently, the annealed samples were digested by T7 Endonuclease I (New England Biolabs) for 30 min and separated by a 2% agarose gel, and quantification was based on relative band intensities. Digitalized images were analyzed to calculate indel efficiency using the Image J software. Indel percentage was determined by the formula:
100 × [1 – √1 – (b + c)/(a + b + c)]
where a represents the integrated intensity of the undigested PCR product, and b and c are the integrated intensities of each cleavage product.
Deep sequencing of Cas12a gene editing results in HEK293T cells
HEK293T cells were transfected and harvested as described for assessing activity of Cas12a cleavage. The genomic-region-flanking DNMT1 targets were amplified using a pair-specific primer with sample-specific barcodes added to the ends of the target amplicons (Supplementary File S3). PCR products were run on 1.6% agarose gel and purified using Spin Column (Sangon Biotech, Shanghai, PR China) as per the manufacturer's recommended protocol. Equal amounts of the PCR products were pooled, and samples were sequenced commercially (Genewiz) by paired-end read sequencing using the Illumina HiSeq X Ten platform.
Off-target analysis for FnCas12a, TEXT, LbCas12a, and AsCas12a
To profile the off-targeting effect of FnCas12a, TEXT, LbCas12a, and AsCas12a, the off-target sites were predicted by CRISPR RGEN Tools (Cas-OFFinder). The fragments containing potential off-target sites were amplified and tested by T7E1 cleavage assay. The sequences of crRNA and off-target sites are listed in Supplementary File S4.
Statistics
Statistical significance was calculated using Mann–Whitney tests using GraphPad Prism v8.4.0 (GraphPad Software, Inc., La Jolla, CA). The error bars in all figures show the standard error of the mean (n = 3). p-Values are reported using GraphPad style, where ns indicates not significant, *p > 0.05, *p < 0.05, **p < 0.01 and ***p < 0.001.
Results
Fusion of FnCas12a and 5′-3′ ssDNA exonuclease can enhance editing efficiency in HEK293T cells
Cas12a generates staggered DSB ends, which can be easily combined by Watson–Crick base pairing and thus will favor the accurate NHEJ repair pathway that does not contribute to the overall editing efficiency (Fig. 1A). To increase the gene editing efficiency of FnCas12a, we assume to bias the DSB repair pathway into imprecise NHEJ by fusing an ssDNA exonuclease with FnCas12a that will degrade the perfectly compatible sticky ends (Fig. 1A). To test our hypothesis, we selected six ssDNA exonuclease candidates, including Artemis from human, RecJ and polA-exo from Escherichia coli, and exonuclease from phage T5, phage T7, and phage λ. Each of them was fused to either the N- or C-terminus of FnCas12a as candidate fusions for comparison tests of the editing efficiency (Fig. 1B). T7E1 assay showed that T5 EXO had the most significant effect on improving the indel efficiency of FnCas12a, increasing by 216% at the N-terminus of FnCas12a and 141% at the C-terminus of FnCas12a (Fig. 1C). Although FnCas12a-Artemis and ecRecJ-FnCas12a showed slightly higher indel efficiency than FnCas12a, the rest of the candidates did not improve the editing efficiency of FnCas12a. Considering that N-terminus-fused T5 EXO-FnCas12a exhibited higher indel efficiency and a smaller size than all other candidate fusions, we chose it for subsequent genome editing tests, and hereafter we refer to it as TEXT.
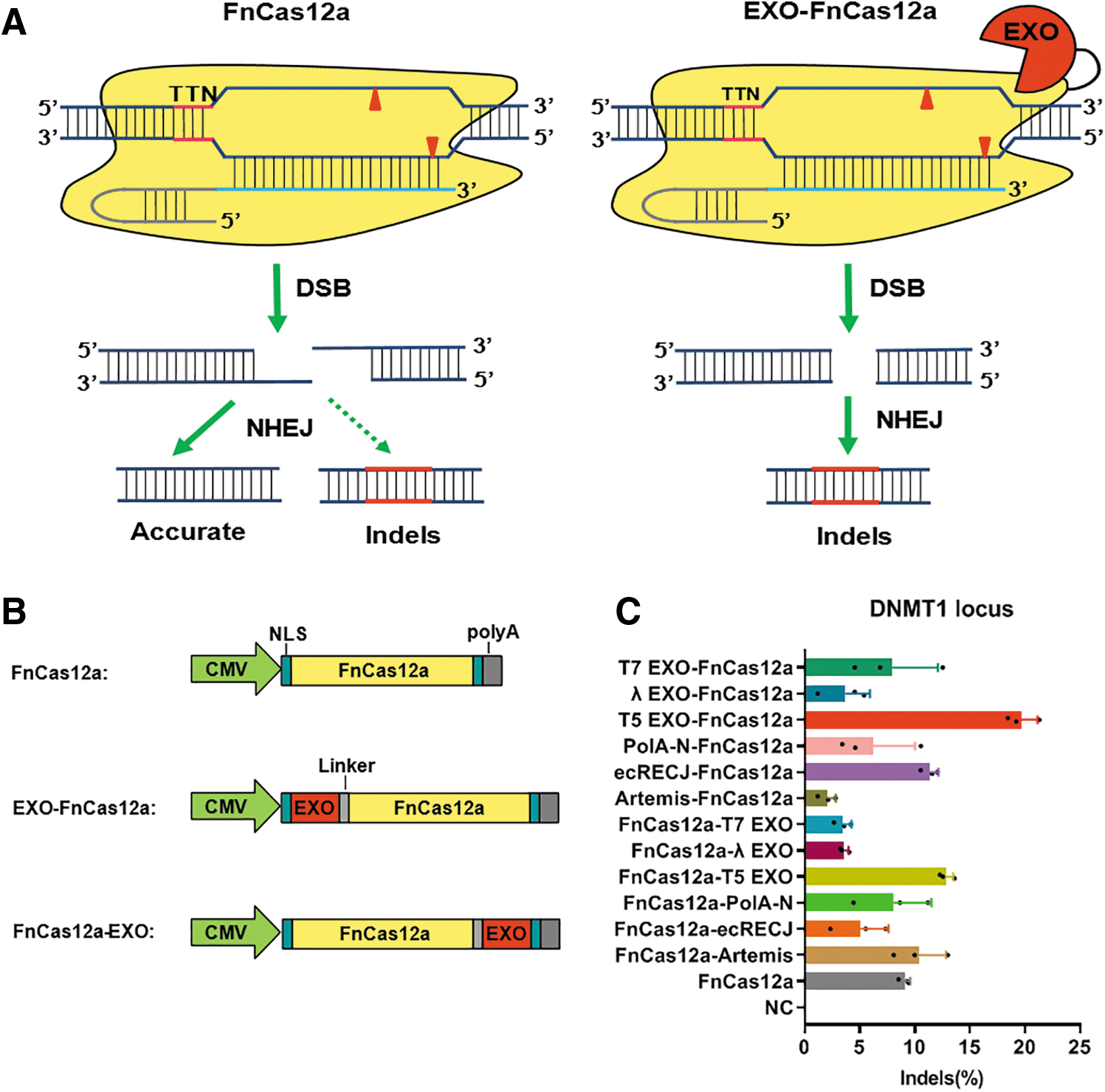
FnCas12a, EXO-FnCas12a, and FnCas12a-EXO mediated gene editing in HEK293T cells.
TEXT genome editing at multiple loci of different cell lines
To profile the TEXT system further, we designed FnCas12a crRNAs for targeting three loci—DNMT1, CCR5, and GAPDH—in different cell lines, including HEK293T cells, Hela cells, and human lens epithelial B3 (HLEB3) cells. Compared to FnCas12a at DNMT1 locus, the TEXT system improved gene editing efficiency by up to 172% in Hela cells and 330% in HLEB3 cells (Fig. 2A). At CCR5 locus, TEXT showed two- to threefold higher editing efficiency than FnCas12a in all three cell lines (Fig. 2B). Notably, at GAPDH locus, FnCas12a only showed around 0.3% editing efficiency in HEK293T cells, and undetectable levels of editing in both Hela and HLEB3 cells, while TEXT dramatically improved gene editing efficiency, with a 10-fold increase in HEK293T cells and with significant editing efficiency in Hela and HLEB3 cells (Fig. 2C). These results indicate that the TEXT system can generally increase gene editing efficiency of FnCas12a in human cells, ranging from 2- to 10-fold higher efficiency in our tests, and can significantly edit locus that previously could not be edited by FnCas12a.
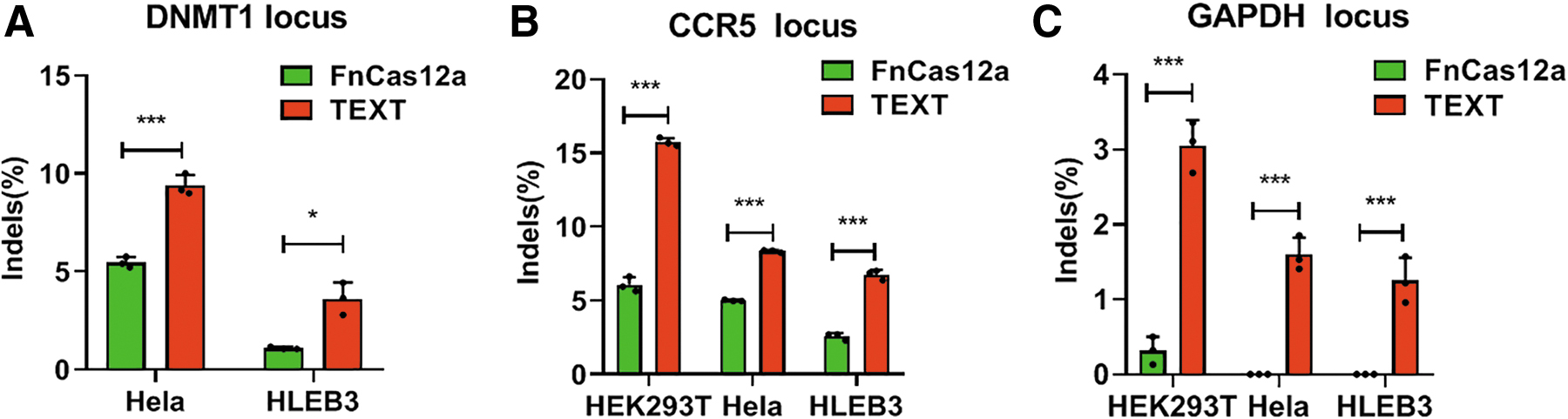
Comparison of gene editing efficiency of the FnCas12a system and the TEXT system in HEK293T, Hela, and HLEB3 cell lines.
Varying spacer length of crRNA to optimize editing efficiency of TEXT
The spacer region of crRNA uses Watson–Crick base paring to locate the Cas12a–crRNA complex or TEXT system at specific genomic loci. It has been reported that by optimizing the spacer length of crRNA ranging from 18 to 23 nt, the cutting efficiency of CRISPR-Cas12a can be increased. Typically, 21 nt can achieve higher editing efficiency than other lengths when using FnCas12a. 16 In a similar fashion, in order to investigate the optimal spacer length of crRNA for TEXT-mediated gene editing in human cells, we constructed a series of crRNAs with different spacer lengths ranging from 18 to 23 nt, and we tested the gene editing efficiency of TEXT at DNMT1 locus. Consistent with a previous report, 16 we found that crRNA with a 20–21 nt spacer length can enable higher gene editing efficiency when using FnCas12a (Fig. 3B), while the TEXT system requires a 21–23 nt spacer length to achieve maximum cutting efficiency (Fig. 3B). In addition, it is worth noting that when the spacer length of crRNA was 18 nt, the editing efficiency of FnCas12a sharply decreased to 1.2%. However, the TEXT system retained more than 10% editing efficiency. Overall, these results confirmed that the TEXT system is compatible with a wide range of guide lengths.
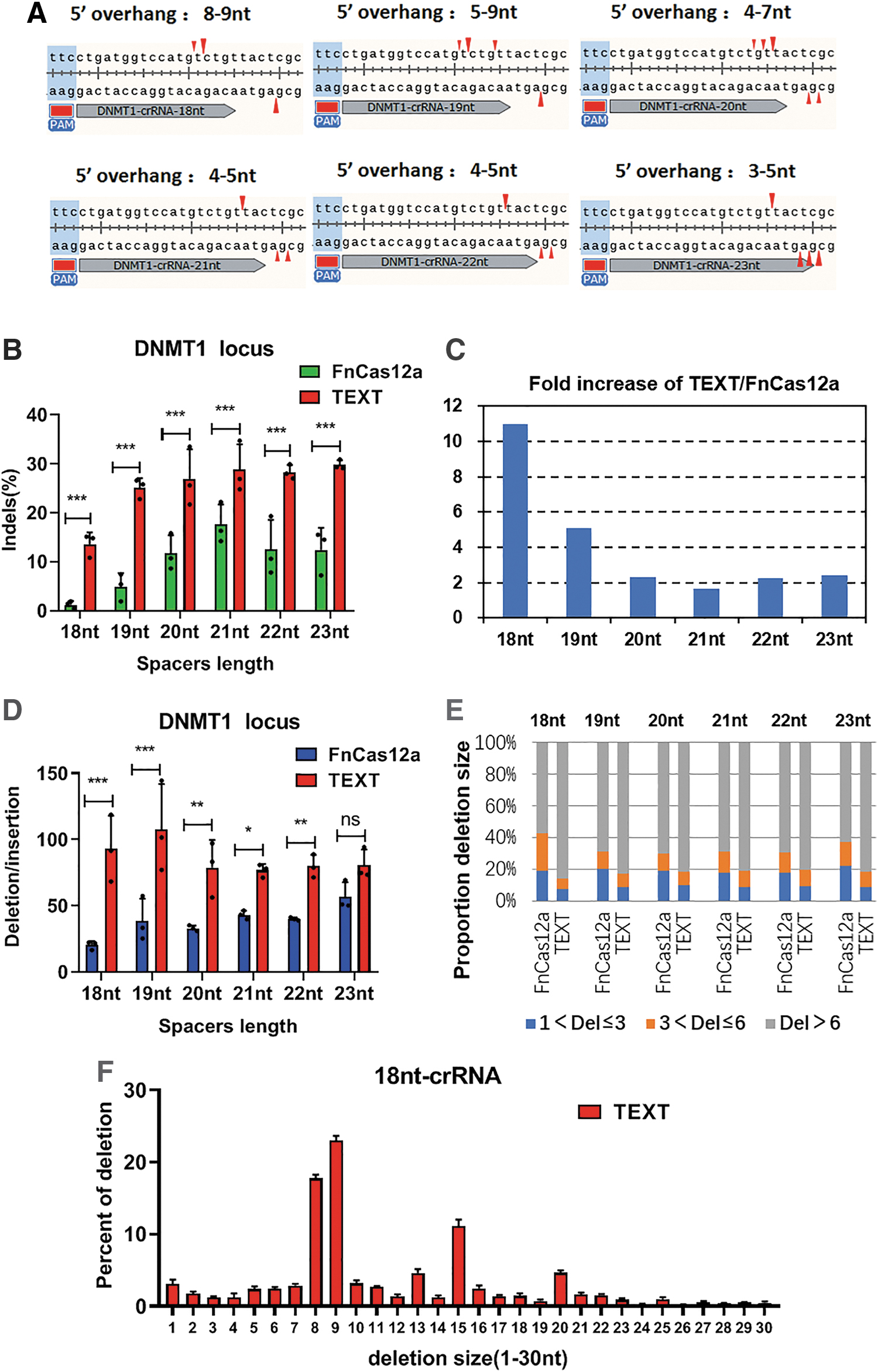
Different spacer lengths of crRNA result in distinct cleavage pattern and gene editing efficiency.
TEXT improves gene editing under different lengths of crRNA
Previously, it was commonly believed that Cas12a would cut DNA at the 18th base downstream of the PAM site on the non-target strand and the 23rd base on the target strand. 9 However, recently, it has been reported that FnCas12a cleavage sites are located after the 13th, 14th, 18th, and 19th bases on the non-target strand and from the 21st to 24th bases on the target strand, 29 which supports the scheme that FnCas12a has a different cleavage pattern when using different spacer lengths of crRNA (Fig. 3A). When the spacer length of crRNA is 18 nt, which typically directs Cas12a to generate 8–9 nt sticky end, the gene editing efficiency of the TEXT system is 11 times higher than FnCas12a (Fig. 3A and C). When the spacer length is 19 nt, which directs Cas12a to generate a 5–9 nt sticky end, TEXT can increase the editing efficiency of FnCas12a by five times (Fig. 3A and C). When the spacer length is 20–23 bp, with 3–5 nt sticky end, the gene editing efficiency of TEXT is around twofold higher than FnCas12a (Fig. 3A and C).
Deep sequencing analysis of TEXT indel patterns
Furthermore, we analyzed the indel pattern of TEXT and FnCas12a with different spacer lengths of crRNA by deep sequencing (Fig. 3D–F). In general, TEXT induced a higher indel efficiency than FnCas12a (Fig. 3B and C). In addition, the ratio of deletion frequency to insertion frequency (del/in) is significantly higher in TEXT than it is in FnCas12a (Fig. 3D). When using 18 nt spacer crRNA, TEXT increased the del/in of FnCas12a from 20.17 to 92.86, while it only increased from 56.78 to 80.84 when using the 23 nt spacer crRNA (Fig. 3D). Then, we measured the lengths of the deletions generated by TEXT and FnCas12a. When using FnCas12a, under different crRNA spacer lengths from 18 to 23 nt, the proportion of deletion sizes <3 bp and <6 bp are about 20% and 30–40%, respectively, while when using TEXT, the deletion sizes that are <3 bp and <6 bp account for 10% and 20%, respectively (Fig. 3E). It is worth noting that when the spacer length is 18 nt, the deletion size that is <6 bp is 42.6% by FnCas12a and 14.3% by TEXT (Fig. 3E). In addition, we analyzed the deletion pattern and found that the proportion of 8–9 nt deletion of TEXT is up to 41.2% with a 18 nt spacer length crRNA, while that of FnCas12a is 9.8% (Fig. 3F). Next, to understand further the role of T5-EXO in TEXT editing, we analyzed next-generation sequencing data to generate detailed indels pattern of TEXT, in which most of the deletion alleles do not contain the sequence of the 5′ overhangs (Supplementary File S5). The results further confirm that the TEXT system improves the gene editing efficiency of FnCas12a by resecting the 5′ single-strand overhang at the DSB ends. Collectively, the TEXT system substantially increased the deletion frequency and deletion size at the targeted locus.
TEXT performs efficient on-target editing with minimal off-target effects
Further characterization of the TEXT system showed substantial editing activities in human cells. We compared the editing efficiency of FnCas12a, TEXT, LbCas12a, and AsCas12a, with NTTV PAMs in human cells. In all cases, we observed higher editing efficiency achieved by TEXT than by FnCas12, LbCas12a, and AsCas12a (Fig. 4A). In particular, TEXT showed robust editing activities on multiple endogenous target sites with ATTV and GTTV PAMs compared to AsCas12a and LbCas12a (Fig. 4A). These results confirm that TEXT recognizes TTV PAM, which will help target genomic sites that are not accessible to LbCas12a and AsCas12a. Next, we determined the off-target effects of TEXT compared to FnCas12a, LbCas12a, and AsCas12a. We used CRISPR RGEN prediction tools to generate a set of potential off-target sites and found that only one off-target site had detectable efficiency by TEXT and other Cas12a orthologs, while most predicted off-target sites showed undetectable off-target effects (Fig. 4B). The results indicated that the TEXT system does not significantly increase off-target effects when enhancing on-target editing efficiency.
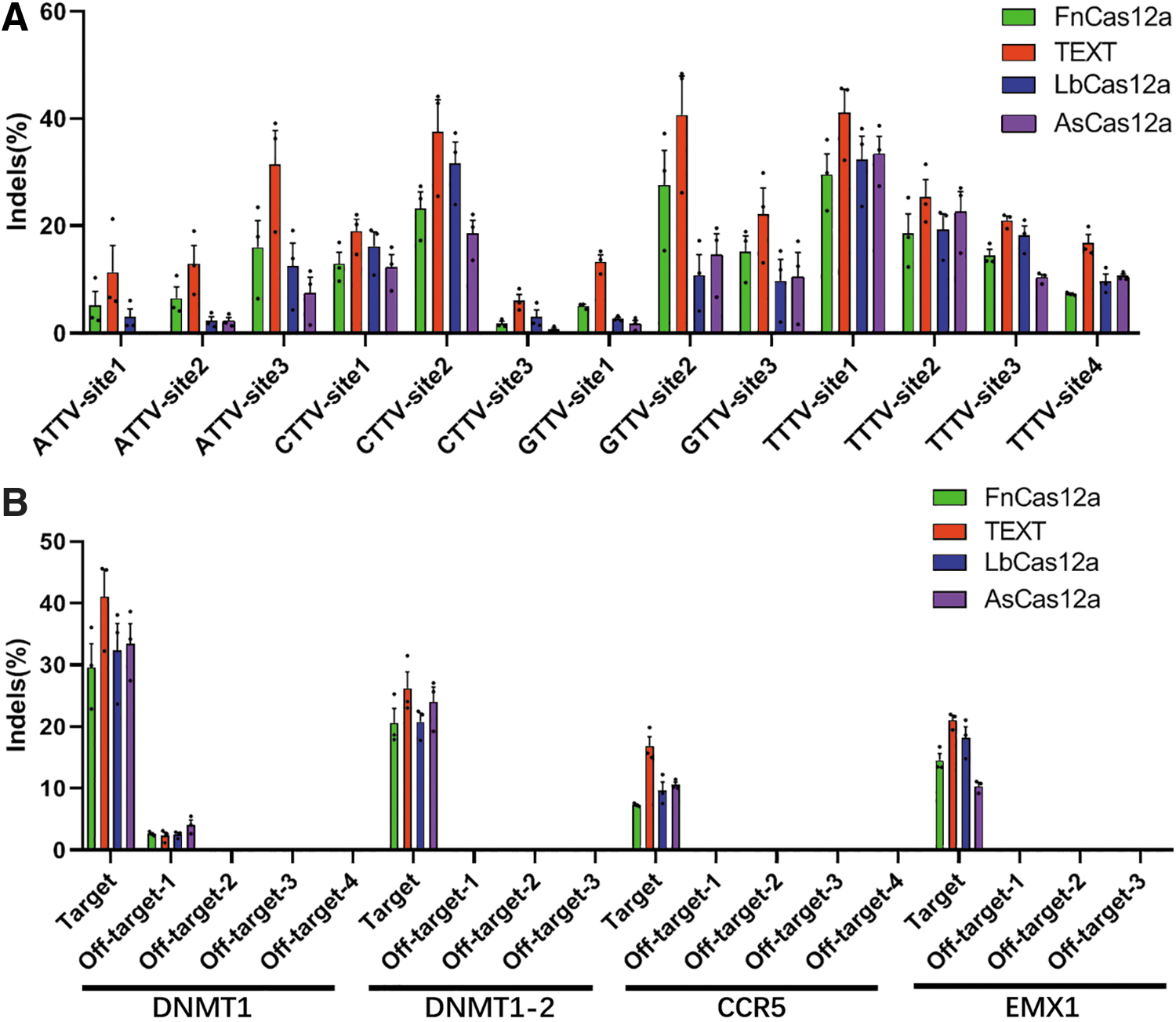
Comparison of editing efficiencies and off-target effects between TEXT and Cas12a orthologs over a diverse range of endogenous targets.
Discussion
Currently, AsCas12a and LbCas12a are more widely used for genome editing than FnCas12a. Although FnCas12a has shown fewer PAM limitations, the low targeting activity of FnCas12a limits its application as a genome editing tool. We report a method that can significantly improve the editing efficiency of FnCas12a by leveraging the endogenous DNA repair pathway to favor the imprecise NHEJ repair that mainly generates indels. We also explored whether our strategy could increase the editing efficiency of LbCas12a and AsCas12a. Unexpectedly, no improvement was observed on those Cas12a variants. A recent report shows that AsCas12a not only generates sticky end, but will also degrade several bases along the non-targeting ssDNA strand, 30 and the structure of LbCas12a and AsCas12a are very similar, which indicates that the nuclease activities of AsCas12a and LbCas12a are high enough to approximate the saturation state of their indel efficiency. Therefore, it is hard to be further improved by tethering T5 exonuclease.
The crRNA of Cas12a consists of a spacer sequence and direct repeat sequence that has a huge impact on the editing efficiency of the CRISPR-Cas12a system.9,21 In vitro experiments have confirmed that different spacer lengths of crRNA can result in different cutting patterns of FnCas12a. 29 We demonstrated that TEXT exclusively improved the gene editing efficiency of FnCas12a when using crRNAs of different spacer lengths (Fig. 3C).
TEXT expands the genome editing activity and scope of FnCas12a with low off-target effects and is superior to AsCas12a and LbCas12a at the tested loci. Our study provides insights into CRISPR-Cas12a systems and provides a new method for maximizing the genome editing efficiency of FnCas12a, enabling it as a promising human genome editing tool for broader research applications. It is worth noting that although no significant cytotoxicity was observed after transfection of the TEXT system, it remains unknown whether exonuclease from phage T5 has detectable off-target effect on naturally occurring DNA damage response in mammalian cells. To understand and improve the security of TEXT further, whole genome sequencing pipelines need to be developed to profile the effect of TEXT comprehensively on naturally occurring DNA lesions. Protein engineering strategies such as rational structural engineering 31 or inducible split protein 32 can be applied to enhance the precision and safety of the TEXT system.
Conclusion
In this study, to improve the editing efficiency of FnCas12a in human cells, we fused T5 ssDNA exonuclease to N terminal of FnCas12a, namely TEXT, which can direct the cellular NHEJ repair more frequently into the error-prone pathway, enabling higher indel efficiency than when using FnCas12a alone. TEXT increases the deletion size under different spacer lengths from 18 to 23 nt, with 18 nt resulting in the highest fold increase. Notably, TEXT shows higher gene editing efficiency than FnCas12a, LbCas12a, and AsCas12a at human genomic loci with NTTV PAMs. In conclusion, we developed an improved FnCas12a system with expanded targeting scope and increased editing efficiency for human cell genome engineering.
Footnotes
Acknowledgments
We thank the facility support of Gene Editing Research Center, Hebei University of Science and Technology. Y. W. and Q.Y. conceived the project, designed all experiments. Y.W., Y.Z., J.S., X.G. and Z.Y. performed the experiments. Y.W. and Q.Y. analyzed the data. Y.W. and Q.Y. wrote the manuscript with help from all authors. Data sets from deep sequencing experiments have been deposited with the National Center for Biotechnology Information Sequence Read Archive under BioProject ID: PRJNA642811.
Author Disclosure Statement
Y.W. and Q.Y. are the inventors on a patent application of this work with aim of ensuring this technology can be used freely and widely.
Funding Information
This work was supported by the Youth Fund of Science and Technology Research Program for Colleges and Universities in Hebei Province (QN2020262).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.