
Abstract
X-linked agammaglobulinemia (XLA) is a monogenic primary immune deficiency characterized by very low levels of immunoglobulins and greatly increased risks for recurrent and severe infections. Patients with XLA have a loss-of-function mutation in the Bruton's tyrosine kinase (BTK) gene and fail to produce mature B lymphocytes. Gene editing in the hematopoietic stem cells of XLA patients to correct or replace the defective gene should restore B cell development and the humoral immune response. We used the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 platform to precisely target integration of a corrective, codon-optimized BTK complementary DNA (cDNA) cassette into its endogenous locus. This process is driven by homologous recombination and should place the transgenic BTK under transcriptional control of its endogenous regulatory elements. Each integrated copy of this cDNA in BTK-deficient K562 cells produced only 11% as much BTK protein as the wild-type gene. The donor cDNA was modified to include the terminal intron of the BTK gene. Successful integration of the intron-containing BTK donor led to a nearly twofold increase in BTK expression per cell over the base donor. However, this donor variant was too large to package into an adeno-associated viral vector for delivery into primary cells. Donors containing truncated variants of the terminal intron also produced elevated expression, although to a lesser degree than the full intron. Addition of the Woodchuck hepatitis virus posttranscriptional regulatory element led to a large boost in BTK transgene expression. Combining these modifications led to a BTK donor template that generated nearly physiological levels of BTK expression in cell lines. These reagents were then optimized to maximize integration rates into human hematopoietic stem and progenitor cells, which have reached potentially therapeutic levels in vitro. The novel donor modifications support effective gene therapy for XLA and will likely assist in the development of other gene editing-based therapies for genetic disorders.
Introduction
Bruton's tyrosine kinase (BTK) is a cytoplasmic kinase that is a lynchpin of multiple signaling pathways, including B cell receptor (BCR) signaling.1–4 Defective BCR signaling halts the development of B lymphocytes and results in the absence of mature B lymphocytes and antibody production, a characteristic of X-linked agammaglobulinemia (XLA).2,5 Without the protection from functional antibodies, patients are susceptible to infections and have reduced life expectancies. 6 The current standard of care for XLA is subcutaneous or intravenous antibody supplementation from healthy donors. This treatment provides a substantial improvement to the patient's quality of life and dramatically increases patient life expectancies.
However, the treatment requires ongoing immunoglobulin injections for life that are expensive and imperfect, leaving susceptibility to recurrent pulmonary infections, invasive viral infections, and inflammatory bowel disease. 6
Allogeneic hematopoietic stem cell (HSC) transplant is currently the only permanent cure for XLA, although it is very rarely performed due to the transplant-associated risks. Graft-versus-host disease and graft rejection are generally considered unacceptably high risks when compared with the moderate severity of clinically managed XLA. Gene therapy using autologous HSCs would maintain the curative effects of allogeneic transplants while reducing the risks. Multiple groups have made progress toward viral vector-mediated gene transfer to deliver a functional BTK gene to human HSCs.7,8
While lentiviral vectors have improved dramatically in recent years, there remains some inherent risk of insertional oncogenesis (IO) with any semirandomly integrating vector.9,10 In some cases, that risk can be tolerated because of the extreme severity of the disease. However, due to the relatively effective current treatment for XLA, any appreciable risk of oncogenesis may be unacceptable. Lentiviral-based XLA therapies have also run into hurdles restoring endogenous expression patterns. Using the natural BTK promoter and enhancer sequences to drive transgene expression produced much lower than wild-type (WT) protein levels. 8 Stronger promoters and enhancers increased this expression, but made it exceedingly difficult to get appropriate expression in all the relevant cell types and may elevate IO risks. 7
It remains somewhat unclear what range of BTK expression is required to restore B cell development and produce protective levels of antibodies. Previous work has demonstrated that BTK expression near physiological levels leads to the most efficient signaling. 11 Overexpression of BTK is correlated with some types of B lymphoid leukemias (e.g., chronic lymphocytic leukemia) and BTK inhibitors such as ibrutinib are revolutionizing treatment for many of these patients.12,13 Although BTK overexpression does not seem to be sufficient for transformation alone, the correlation is worrisome for XLA gene therapies.
These data together suggest that a relatively narrow window of BTK expression will be clinically beneficial; too little BTK expression may not restore B lymphopoiesis, while too much may affect signaling efficiency or even carry risks of oncogenesis. Our approach for correcting XLA instead utilizes the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 platform to improve the fidelity of treatment by first creating a targeted double-stranded DNA break (DSB) at the BTK locus. 14
Following Cas9-mediated DNA cleavage at the target site, the cell can use one of multiple mechanisms to repair the DSB. The most notable of these pathways are nonhomologous end joining (NHEJ), which results in deletions or insertions of random nucleotides at the repair site, or homologous recombination using a template DNA molecule to guide repair, which is the basis of this method of gene therapy. Homology-directed repair (HDR) of BTK mutants can occur if high numbers of a corrective BTK donor DNA are present in the nucleus during DSB repair. These donor molecules contain the BTK complementary DNA (cDNA) sequence flanked by “homology arms” that parallel the cut site and serve as templates for homologous recombination.15,16
While other genetic diseases may feasibly be treated by reverting pathogenic mutations directly, the wide spread of potentially pathogenic mutations throughout the BTK gene makes this approach impractical to cover the majority of patients in need. Instead, addition of a corrective copy of the BTK gene into the start of the gene could be an effective treatment for every patient with exonic mutations anywhere downstream of the target site.
We utilized the CRISPR-Cas9 to integrate a potentially therapeutic, human BTK cDNA sequence into the 5′ end of the endogenous BTK locus. We initially observed suboptimal BTK protein production from the wild-type cDNA and identified several modifications to the BTK transgene cassette to dramatically improve expression levels. Integration and expression from donor integration at multiple target sites were assessed and optimized to produce a novel therapy that may provide a safe, effective gene therapy for XLA.
Materials and Methods
Donor template assembly
A human BTK cDNA was synthesized with codon optimization via the GeneOptimizer web tool (Thermo Fisher Scientific, Waltham, MA) and commercially synthesized by IDT (Integrated DNA Technologies, Coralville, IA). All of the donor templates contain BTK cDNA exons 2 through 19 (2010 bp), the BTK 3′ untranslated region (UTR) (428 bp), three C terminal hemagglutinin (HA) epitope tags attached by a linker (99 bp), and two homology arms that match the sequences flanking the respective target site in the genomic DNA (500 bp each). Donors for the intron 1 target site also included a small portion of the intron 1 sequence (33 bp) between the cut site and the start of exon 2, to ensure efficient splicing.
Guide RNA and Cas9
For K562 and Jurkat experiments, single gRNA was delivered via the pX330-U6-Chimeric_BB-CBhhSpCas9 expression plasmid, which was a gift from Feng Zhang (Addgene plasmid #42230; http://n2t.net/addgene:42230; RRID: Addgene_42230). 14 Paired oligonucleotides representing the gRNA sequences were synthesized (Integrated DNA Technologies), annealed, and cloned into the gRNA/Cas9 expression plasmid. For experiments with gRNA delivered as RNA (rather than on an expression plasmid), it was generated via in vitro transcribed (IVT) or chemically synthesized (Synthego Corporation, Menlo Park, CA) as specified in each experiment. IVT guide RNA was produced from a polymerase chain reaction (PCR)-generated template using the HiScribe T7 Quick High Yield RNA Synthesis Kit (Cat: E2050S; New England Biolabs, Ipswich, MA). 17 Chemically synthesized gRNA was purchased as either unmodified or chemically modified with 2′-O-methyl and 3′ phosphorothioate internucleotide linkages on the three terminal residues (Synthego Corporation). 18 All Cas9 used in this work were Streptococcus pyogenes Cas9 (SpCas9) with the exception of the high-fidelity engineered variants. Cas9 mRNA was also produced via IVT. A plasmid containing wild-type Cas9 was cloned with the protein under the control of the T7 promoter. The plasmid was linearized via restriction digest and the IVT was performed using the mMESSAGE mMACHINE T7 Ultra Transcription kit (Cat: AM1345; Thermo Fisher Scientific). The resulting mRNA was purified using the RNeasy MinElute Kit (Cat: 74204. Qiagen, Germantown, MD). Cas9 proteins for wild-type SpCas9, VP12 Cas9, and eSpCas9 were obtained from the University of California, Berkeley QB3 MacroLab.19,20 Alt-R HiFi Cas9 protein was purchased from IDT (Cat: 1081060; Integrated DNA Technologies). 21
Electroporation of cell lines
K562, Jurkat, and Ramos cells were cultured in “R10”: RPMI 1640 (Cat: 15-040-CV; Corning, Corning, NY) supplemented with 1 × penicillin/streptomycin/glutamine (Cat: 10378016; Thermo Fisher Scientific) and 10% fetal bovine serum (Omega Scientific, Tarzana, CA). Each cell type required different electroporation conditions. K562 and Jurkat cell electroporations were carried out at 2e5 cells per condition. Cells were spun at 90 g for 10 min, the supernatant was aspirated, and the cell pellet was resuspended in Lonza buffer (SF buffer for K562, SE buffer for Jurkat) at 20 μL/condition (Lonza, Basel, Switzerland). The resuspended cells were transferred to prealiquoted tubes containing the payload to be delivered into the cells. For plasmid delivery, 500 ng of pX330 sgRNA/Cas9 expression plasmid was used with 3 μg of BTK donor template plasmid in the TOPO 2.1 plasmid backbone. With endonucleases delivered as ribonucleoprotein (RNP), 200 pmol of Cas9 was added to 9 μg of single-guide RNA (sgRNA) and incubated at room temperature for 15 min before adding to cell mixtures. The resulting reaction mixture was electroporated using the Lonza 4D Nucleofector system (FF120 protocol for K562, CL120 for Jurkat) and allowed to rest for 10 min before plating in 480 μL of R10 medium (described above) per sample. Ramos cell line electroporations were carried out using the Invitrogen Neon electroporation system (Cat: MPK5000; Thermo Fisher Scientific). For the Ramos cells, 7.6 × 104 cells per condition were resuspended in resuspension buffer R (Thermo Fisher Scientific). gRNA, 2.25 μg, and 50 pmol of Cas9 protein were precomplexed for 10 min and added to the resuspension mixture. Samples were electroporated at 1350 V, 30 ms, 1 pulse before being plated in 250 mL of R10 containing the relevant adeno-associated virus (AAV) donor. One day postelectroporation, the medium was changed and the transducing vector removed.
Electroporation/transduction of human CD34+ cells
Granulocyte colony-stimulating factor (G-CSF)-mobilized peripheral blood CD34+ cells (HemaCare, Van Nuys, CA) were prestimulated for 2 days in X-Vivo 15 medium (Cat: BEBP04-744Q; Lonza) supplemented with 1 × penicillin/streptomycin/glutamine (Cat: 10378016; Thermo Fisher Scientific), 50 ng/μL recombinant human (rh) stem cell factor (Cat: 300-07; PeproTech, Rocky Hill, NJ), 50 ng/μL rh Flt-3 ligand (Cat: 300-19; PeproTech), and 50 ng/μL of rh thrombopoietin (Cat: 300-18; PeproTech). Cells, 2 × 105, were resuspended in 100 μL of BTXpress electroporation solution (Cat: 45-0802; BTX, Holliston, MA) and electroporated using the BTX ECM 830 Square Wave Electroporator set to 255 V, 5 ms, and 1 pulse (Cat: 45-0661; BTX). Five micrograms of gRNA and 5 μg Cas9 mRNA or 200 pmol of Cas9 protein were used, as specified by the delivery method in each figure. Following electroporation, cells were left to rest for 10 min in the supplemented X-Vivo 15 culture condition described above before a 24-h transduction with the adeno-associated virus serotype 6 (AAV6) donor vector custom ordered from Virovek (Virovek, Hayward, CA) or Vigene (Vigene Biosciences, Rockville, MD). After transduction, cells were counted to determine the viability and fold expansion before being replated in outgrowth media: Iscove's modified Dulbecco's medium (Cat: 12440053; Thermo Fisher Scientific), 10% fetal bovine serum, 1 × penicillin/streptomycin/glutamine (Cat: 10378016; Thermo Fisher Scientific), 5 ng/mL of rhIL-3, 10 ng/mL of rhIL-6, and 25 ng/mL of rhSCF (Cat: 200-03, 200-06, and 300-07; PeproTech). The use of anonymous medical waste samples of CD34+ hematopoietic stem cells has been deemed not human subjects research and is IRB exempt at UCLA.
Measuring allelic disruption
A 638 bp amplicon that encompasses BTK exon 2 as well as portions of introns 1 and 2 was amplified from genomic DNA taken from edited cells (primer sequences: “GCCATTTAACATCTAGAGCATTCC,” “GTGGCTTCTTAGGACCTTTGAC”). The resulting amplicon was Sanger sequenced and analyzed via the Synthego ICE analysis web tool (Synthego Corporation, Menlo Park, CA).
GUIDE-seq
GUIDE-seq (genome-wide unbiased identification of double-stranded breaks enabled by high-throughput sequencing) was performed in K562 cells evaluating the specificity of the sgRNA as described by Kuo et al. 22
Integration analysis
Genomic DNA was extracted from edited cells for integration site analysis using the Invitrogen PureLink Genomic DNA Kit (Cat: K182002; Thermo fisher Scientific) and quantified using the NanoDrop system (Cat: ND-2000; Thermo Fisher Scientific). DNA samples were then analyzed by droplet digital PCR to measure integration rates. Two sets of primers were duplexed, each with their own fluorescent probe (FAM/HEX). One primer was complementary to a BTK gene sequence upstream from the left homology arm of the donor. The second primer bound to the codon-optimized BTK donor sequence, to allow for specific measurement of the integrated BTK transgene distinct from the endogenous BTK gene. The FAM-conjugated nucleotide probe with a quencher also bound to the minus-strand DNA near the second primer.
A reference primer/probe set was also delivered to recognize the UC462 region of the X chromosome. One microliter of EcoRV-HF endonuclease (Cat: R3195S; New England Biolabs) was added to the reaction mixture (an enzyme that does not disrupt the experimental or reference amplicons) to reduce background. Each sample was digested at 37°C for 1 h before droplet generation with the Bio-Rad QX200 Droplet Generator (Cat: 186-4002; Bio-Rad, Hercules, CA). The prepared samples were then assayed via the QX200 Bio-Rad Droplet Reader on the “Absolute” measurement setting (Cat: 186-4003; Bio-Rad).
BTK phosphorylation in Ramos cells
A total of 2 × 106 Ramos cells per condition were stimulated with 10 μg/mL of goat F(ab′)2 specific to human IgM (Cat: H15100; Thermo Fisher Scientific). The cells were incubated at 37°C for 30 min before protein lysates were prepared using the RIPA lysis and extraction buffer (Cat: 89900; Thermo Fisher Scientific) supplemented with 1 × HALT protease and phosphatase inhibitor cocktail (Cat: 78444; Thermo Fisher Scientific).
Immunoblot analysis
For immunoblots probing for nonphosphorylated proteins, cells were lysed in denaturing cell extraction buffer (Cat: FNN0091; Thermo Fisher Scientific) with added HALT protease inhibitor (Cat: 87786; Thermo Fisher Scientific) at a 1 × concentration following the manufacturer's protocols. Cells were first spun at 500 g for 5 min at 4°C, the supernatant was aspirated, and the cell pellet was washed with chilled Dulbecco's phosphate-buffered saline (Cat: 14190250; Thermo Fisher Scientific). The spin, aspirate, and wash steps were repeated. Then, chilled DCEB+HALT was added to the cell pellet at a concentration of 100 μL per 2e6 cell and incubated at 4°C for 30 min, vortexing every 10 min. The samples were then spun at 17,000 g for 15 min at 4°C before transferring the supernatant to a fresh tube. Processing lysates for analysis of phosphorylation sites used the same procedure except lysis was performed using RIPA buffer with HALT protease and phosphatase inhibitor being added (Cat: 78440; Thermo Fisher Scientific).
Lysate concentrations were determined using the Pierce BCA protein assay (Cat: 23227; Thermo Fisher Scientific) following the manufacturer's protocol. Samples were treated for sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) with NuPAGE LDS Sample Buffer (Cat: NP0007; Thermo Fisher Scientific) and NuPAGE Sample Reducing Agent (Cat: NP0009; Thermo Fisher Scientific), each to a 1 × concentration. Lysates were diluted to contain equivalent amounts of integrated BTK gene copies for immunoblot gel loading, using lysate from the BTK-deficient K562 cells to keep constant the total amount of protein loaded per lane to allow for valid loading controls. Wild-type K562 lysate was used as a control to indicate the relative expression levels of the edited cells compared with normal cells. Wild-type lysate was diluted similarly to match its copy level of its endogenous BTK gene, the inserted BTK gene copies, and contain a constant total protein amount.
The data table below illustrates the calculations described here.
These samples were heated to 95°C for 10 min before loading into a 4–12% Bis-Tris gel (Cat: NP0322BOX; Thermo Fisher Scientific). Samples were transferred to polyvinylidene fluoride transfer membrane (Cat: 88520; Thermo Fisher Scientific). The membrane was blocked with 5% milk in 1 × Pierce TBS Tween 20 buffer (Cat: 28360; Thermo Fisher Scientific) for 1 h. Membranes were split into discrete portions to probe for different proteins and incubated in a 1:1000 dilution of primary antibody for BTK (Cat: 56044S; Cell Signaling Technology), phosphorylated BTK (Cat: 5082S; Cell Signaling Technology), HA (Cat: NBP2-43714; Novus, Irvine, CA), or actin (Cat: 3700S; Cell Signaling Technology), shaken overnight at 4°C.
Membranes were washed three times with TBS Tween and incubated in 5% milk with a secondary antibody for 90 min (Cat: 554002; BD Biosciences, San Diego, CA) (Cat: A-21236; Thermo Fisher Scientific). Secondary antibodies were washed off three times. Blots stained with horseradish peroxidase-conjugated antibodies were treated with Pierce ECL Plus Western Blotting Substrate (Cat: 32132; Thermo Fisher Scientific). All blots were imaged via phosphorImager and quantified via densitometry.
Quantifying RNA abundance via reverse transcription digital droplet PCR
Total RNA was extracted from treated cells, 1e6 cells per sample, with the RNeasy Mini Plus Kit (Cat: 74136; Qiagen) following the manufacturer's protocol. Purified RNA was transferred to 0.2 mL PCR tubes with 0.1 volume 10 × Turbo DNase buffer (Cat: AM1907; Thermo Fisher Scientific) and incubated at 37°C for 30 min. We used 0.1 volume of DNase Inactivation Reagent incubated at room temperature for 5 min and then centrifugated the samples at 10,000 g for 1.5 min taking the supernatant. cDNA was synthesized from purified DNase-treated RNA with the SuperScript III First-Strand Synthesis System for reverse transcription (RT)-PCR (Cat# 18080-051; Invitrogen) following the manufacturer's protocol.
To analyze mRNA expression of the BTK transgene donor, primers were designed to be specific to the codon-optimized construct and the WT sequence. IPO8 was chosen as the reference gene. The cDNA were diluted 25-fold and gene expression was determined by RT-digital droplet PCR (ddPCR).
Results
A BTK cDNA integrated efficiently into the endogenous locus but produced subphysiological levels of BTK expression
Initially, a Cas9 target site at the 3′ end of BTK intron 1 was chosen because it could be curative to patients with any loss-of-function mutations in the BTK protein coding region (Fig. 1a). Multiple-guide RNAs were tested in cell lines to identify the optimal site for allelic disruption (data not shown). A donor plasmid was designed to achieve targeted integration of the full BTK cDNA sequence to the DSB created by the Cas9/sgRNA at this site.

Targeted integration of BTK cDNA into intron 1 of the gene leads to subphysiological levels of mRNA and protein expression.
This BTK sequence was codon optimized to reduce homology to the intact genomic locus without changing the amino acids encoded, to reduce the potential for unwanted integration events (Supplementary Fig. S1). 23 Three methods of codon optimization were tested for integration and expression rates, with sequence optimized via the GeneOptimizer web tool producing the highest efficiency (data not shown). Three C-terminal hemagglutinin tags were added upstream of the stop codon to differentiate between wild-type (endogenous) and transgenic BTK. The full BTK 3′ UTR was also included following the stop codon to support transcriptional termination and polyadenylation. Five hundred base-pair long homology arms were added to either side of the donor construct that matched the sequences flanking the target site in the genomic DNA.
The sgRNA/Cas9 was delivered as an expression plasmid in conjunction with a plasmid containing the described BTK donor. BTK-deficient K562 cells were created using Cas9 to generate a homozygous 4 bp deletion in exon 15 of the BTK gene and the absence of BTK protein was verified via immunoblot (Supplementary Fig. S2).
Delivery of the Cas9/sgRNA endonuclease alone to the BTK-deficient K562 cells generated allelic disruption in 24.3% ± 5.1% of cells (Fig. 1b). Adding only the Cas9/sgRNA endonuclease plasmid or only the BTK cDNA donor plasmid led to no detectable targeted integration, while adding both nuclease and donor together achieved integration in 13.0% ± 1.1% of cells (Fig. 1c). Immunoblots probing for the HA tags on the BTK donor demonstrated that the transgenic protein was present in the BTK-disrupted K562 cells (Fig. 1d). However, probing for the BTK protein showed that the actual expression per cell was only 11.1% ± 1.4% of the wild-type protein in parental K562 cells (Fig. 1d). Transgenic BTK mRNA normalized for the DNA integration rate was only at 20.2% ± 1.5% of wild-type levels (Fig. 1e).
To identify off-target cleavage events, GUIDE-seq was performed in K562 cells and detected only on-target cleavage events with this sgRNA to BTK intron 1 (Fig. 1f). 24
To test for function of the transgenic BTK construct, Ramos cells served as a better model than K562 cells because they are a B lymphocyte cell line. The presence of a BCR in Ramos cells allows for direct assessment of the signaling pathway aiming to be restored. A Ramos cell subline was engineered using Cas9-mediated gene knockout to have a frameshift mutation in BTK exon 15 that led to an early stop codon, (data not shown).
The BTK-deficient Ramos line was then used to create a clonal BTK rescue line with the Cas9/sgRNA and BTK cDNA donor described above integrated into the intron 1 site. Wild-type, BTK-deficient, and BTK-rescued (gene edited) Ramos clonal cell lines were stimulated with a BCR-crosslinking antibody fragment and immunoblotted for phosphorylated BTK (Fig. 1g). The BTK-rescued cell line responded to the BCR-crosslinking with an increase in BTK protein phosphorylation, suggesting that the transgene folds and localizes correctly.
Addition of the BTK terminal intron or the Woodchuck hepatitis virus posttranscriptional regulatory element to BTK donor templates boosted transgene expression
Previous work identified the importance of introns for optimal transgene expression.25,26 Multiple mechanisms function together for this phenomenon; intron splicing improves nuclear export of the transcript and polyadenylation of the nascent RNA. 27 In addition, introns can include important enhancer sequences. The terminal intron of a gene plays a particularly important role in efficient transcription termination and 3′ end processing of transcripts; removal of the 3′ splice site immediately preceding the terminal exon led to a substantial drop in polyadenylation of the resulting transcript. 28
The terminal intron of BTK, intron 18, is 3.2 kb long, which would be too large to package with the rest of the BTK cDNA donor in an AAV6 vector. 29 These AAV6 vectors are ideal for efficient donor delivery into primary cells, while plasmids yield very high toxicity with low efficacy in hematopoietic stem and progenitor cells (HSPCs). 30 However, the donor can still be tested in cell lines via delivery as a plasmid. To the best of our knowledge, BTK intron 18 has no known or putative enhancer binding sites and no patients have been identified with pathogenic mutations solely in intron 18 (except mutations that disrupted splice sites).
Three donor variants were created: one with the full intron 18 (I18), another with a 679 bp truncated variant of intron 18 (I18t), and finally, one with the much smaller intron 6 of BTK intron (I6), each inserted between exons 18 and 19 of the cDNA sequence. Importantly, all three variants retained the 5′ and 3′ splicing signals (Fig. 2a). These donors, as well as the intronless base donor (“cDNA”), were assessed as electroporated plasmids in BTK-deficient K562 cells.
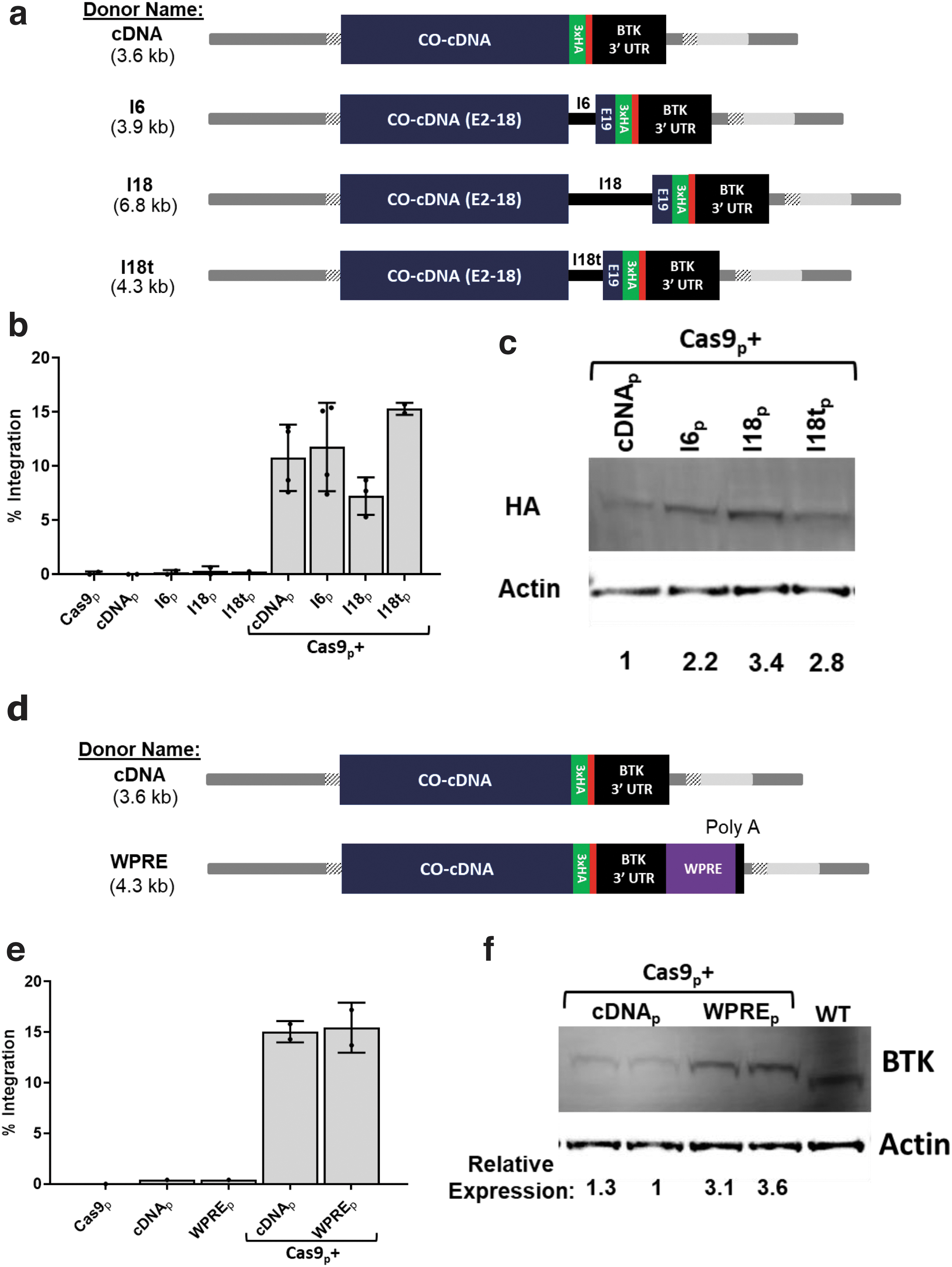
Addition of a terminal intron or the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) to donor templates increases transgenic BTK expression.
The full intron 18 donor generated the lowest rates of integration at 7.2% ± 1.7%, while the other three donors had similar integration frequencies between 10.8% and 15.3% (Fig. 2b). The presence of any of the three intron variants led to increased BTK expression per modified cell (Fig. 2c). The donors containing the I6, I18, and I18t intron variants produced 2.2, 3.4, and 2.8 times as much of the transgenic protein as the base cDNA donor. The I18t donor markedly increased BTK expression while remaining of a size compatible with packaging into AAV6 vectors.
Another element worth assessing for the potential to increase expression levels is the Woodchuck hepatitis virus posttranscriptional regulatory element (WPRE). WPRE is often added to retro- and lentiviral vectors to improve titers by increasing the abundance of the viral RNA transcript in the packaging cells. 31 It is also used in some types of vectors to increase transgene expression levels. This element has been used for decades, although there remains some degree of uncertainty about its mechanisms of action. The most commonly described mechanism of action is its formation of a tertiary structure, sterically blocking the 3′ end of the mRNA strand from degradation. Other proposed mechanisms include improved transcriptional terminal or more efficient nuclear export of transcripts.
We designed a new BTK cDNA donor variant for integration into BTK intron 1 with the WPRE added immediately after the 3′ UTR, just before the polyadenylation signal (Fig. 2d). This new WPRE containing a donor was tested head to head with the base cDNA donor in BTK-deficient K562 cells. While integration rates were similar between the two donors, the WPRE-containing BTK donor produced nearly tripled BTK protein expression (Fig. 2e, f).
Simultaneous addition of a protospacer adjacent motif mutation, a truncated intron, and the WPRE to BTK donor templates produced nearly wild-type levels of BTK expression
While the codon optimization of exonic donors usually disrupts the sgRNA binding site, intronic regions such as the BTK intron 1 site have no codons to be optimized, and so, the full Cas9 binding site and protospacer adjacent motif (PAM) sequence remain intact. The preservation of this Cas9/sgRNA binding site allowed the integrated donor to be recleaved and repaired via NHEJ, generating indels at the integration junction of every sample tested (data not shown). Removing the PAM sequence within the BTK donor (ΔPAM) resulted in 100% base perfect 5′ integration junctions without affecting the total integration rates in K562 cells.
Donor plasmids were designed containing combinations of the three modifications described: ΔPAM, truncated intron 18 addition, and WPRE addition. When both the truncated intron and the WPRE were added to the donor plasmid, the resultant size was too large to be efficiently packaged into an AAV vector, necessitating the creation of a further truncated intron 18 variant, dubbed microintron 18 (I18u). The new variants created were as follows: ΔPAM-WPRE, ΔPAM-I18t-WPRE, and ΔPAM-I18u-WPRE (Fig. 3a). Each of these donor templates were electroporated as plasmid into BTK-deficient K562 cells in conjunction with the BTK intron 1 targeting sgRNA/Cas9 expression plasmid. The integration rates of each donor were comparable except the base ΔPAM donor, which integrated at slightly higher efficiency (Fig. 3b).
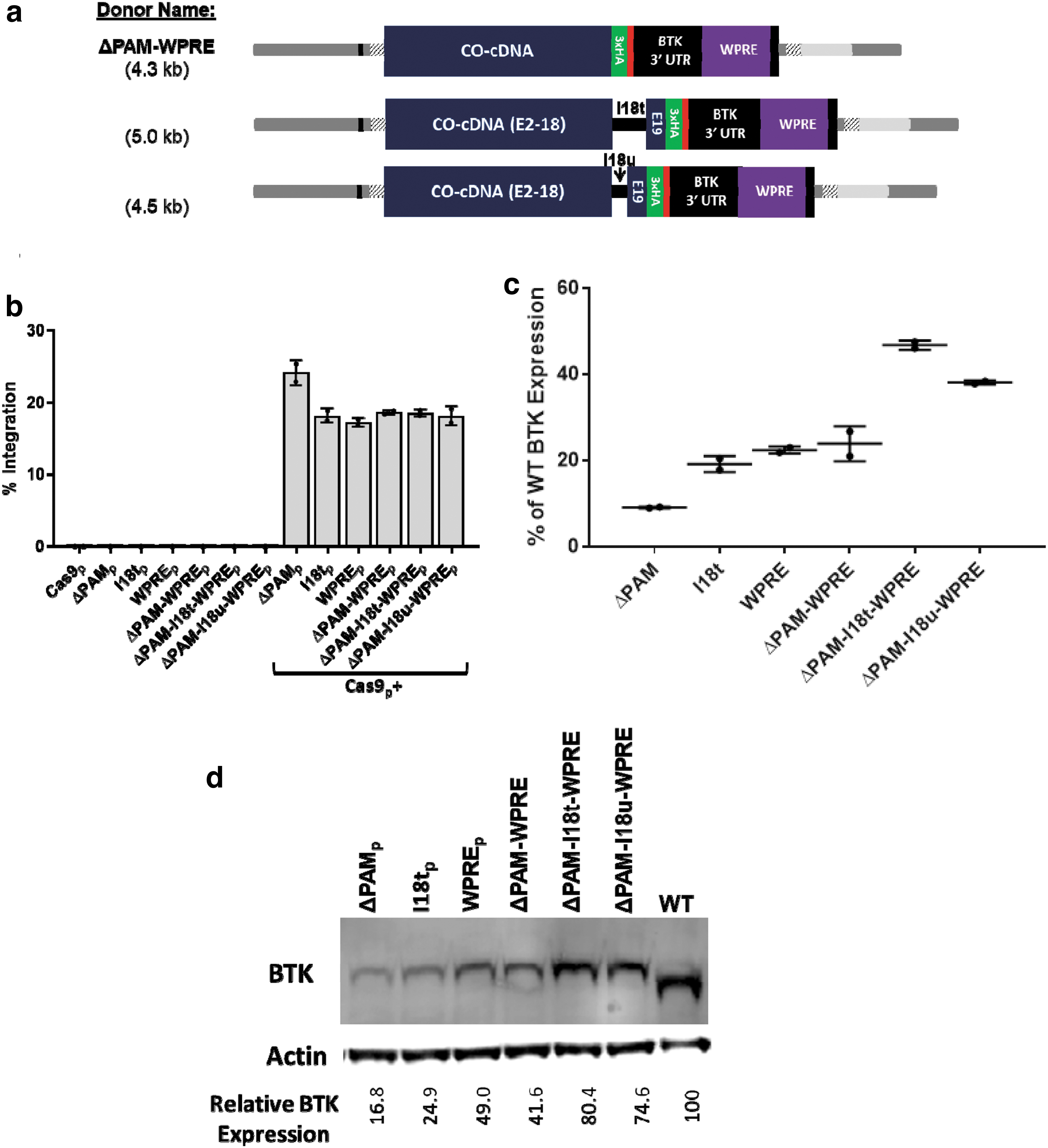
Combining successful donor modifications leads to an additive effect on transgene expression.
RNA from these cells was collected and analyzed via RT-ddPCR to determine relative quantities of the BTK transcripts (normalized to the housekeeping gene IPO8). This expression was also scaled via the previously determined integration rates to effectively compare expression per integrated (or wild type) copy of the BTK gene. The base ΔPAM donor produced only 9.1% ± 0.2% of wild-type expression (Fig. 3c). The I18t donor generated 19.2% ± 1.9% of wild-type levels, while WPRE alone led to 22.4% ± 0.8% and 23.9% ± 4.1% with the PAM mutation. Combining the three elements led to the highest expression—46.8% ± 1.1% of wild-type BTK. The packageable combination of the elements (ΔPAM-I18u-WPRE) generated second highest RNA levels at 38.1% ± 0.5%.
Protein lysates from each treated population were prepared and analyzed via immunoblot for BTK, with actin as a loading control. The results reflected the trend seen in the RNA expression data (Fig. 3d). The base donor produced 16.8% the amount of BTK protein per copy of the gene when compared with wild-type cells. Integration of the I18t containing donor led to an increase in expression to 24.9% of wild type. Donors with the WPRE yielded 49.0% of wild-type expression with the PAM intact and 41.6% for the ΔPAM-WPRE variant. Again, the highest expression came from the ΔPAM-I18t-WPRE donor at 80.4% of wild type, while the packageable-sized variant (ΔPAM-I18u-WPRE) performed nearly as well, with 74.6% of wild-type expression.
Addition of the WPRE, but not the truncated intron 18, produced elevated BTK expression in a T lymphocyte line
While the integration of a donor with both the truncated intron 18 and WPRE led to nearly wild-type levels of BTK mRNA and protein, the inclusion of these elements raised the question of whether the lineage specificity of the donor was maintained. While BTK is found in most hematopoietic lineages, there are notable exceptions such as T lymphocytes. 32 Four BTK donors were compared in the Jurkat T cell line by electroporation of the Cas9/sgRNA and donor plasmids: ΔPAM, I18t, ΔPAM-WPRE, and ΔPAM-I18u-WPRE. Both WPRE-containing donors yielded about three times higher rates of integration than the donors lacking the WPRE (Fig. 4a). Immunoblot analysis of these treated cells normalized for percent integration demonstrated BTK protein expression from Jurkat T cells treated with the WPRE-containing donors, but not from the base or truncated intron 18 donors (Fig. 4b).
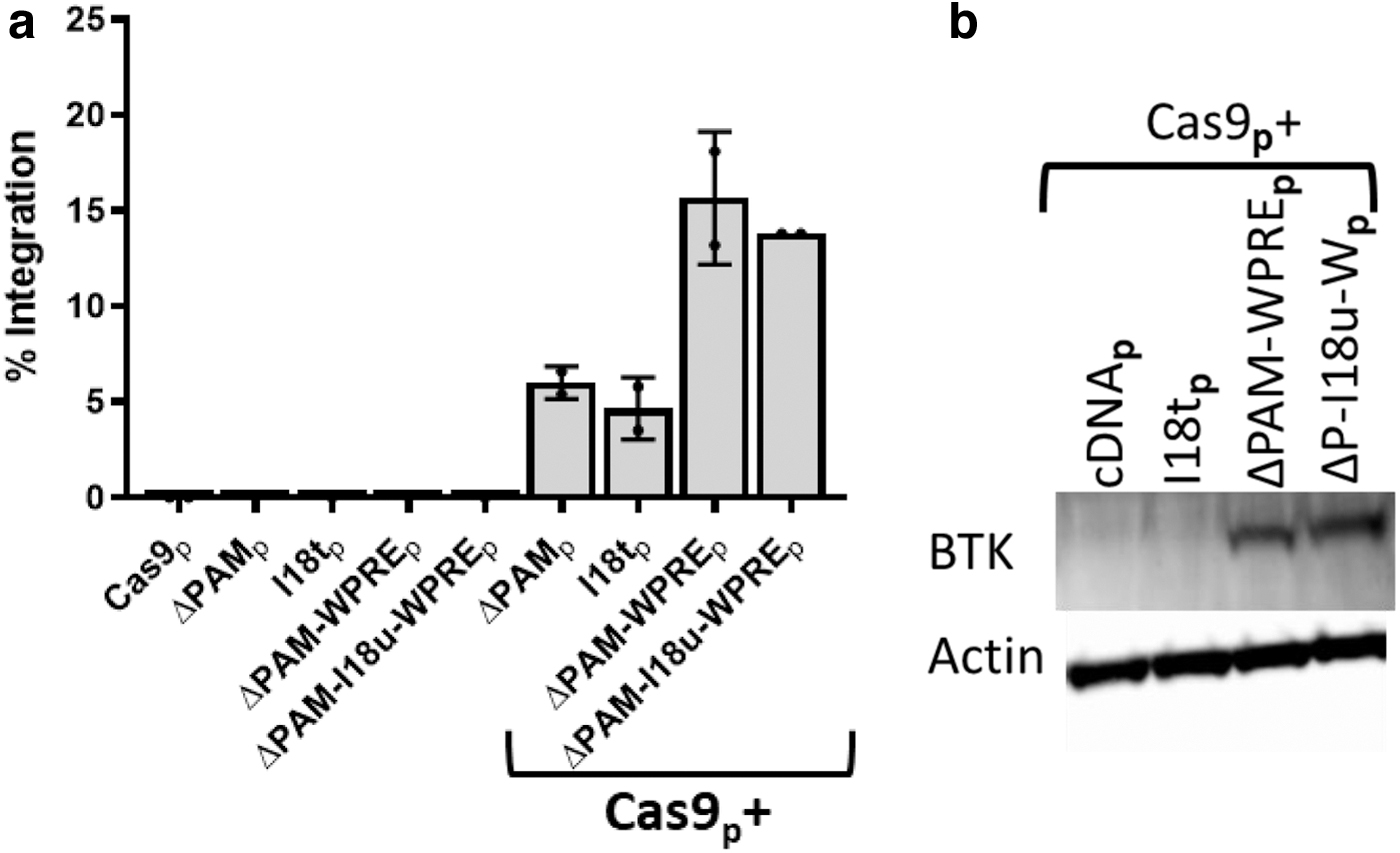
Integration of WPRE containing donors led to elevated BTK expression in non-BTK expressing Jurkat T lymphocytes.
Optimization of donor integration into BTK intron 1 in human CD34+ HSPCs
Primary human G-CSF-mobilized peripheral blood CD34+ HSPCs were thawed and prestimulated for 2 days before electroporation with the sgRNA/Cas9. For experiments assessing integration rates, cells were transduced with an AAV6 vector containing the ΔPAM donor for 24 h immediately following electroporation (Fig. 5a). Cas9 was delivered as either an RNP precomplexed with the sgRNA or as Cas9-encoding mRNA. The sgRNA was either IVT or chemically synthesized with 2′-O-methyl and 3′ phosphorothioate internucleotide linkages on the three terminal residues on both the 5′ and 3′ ends of the molecule. These chemical modifications have been reported to increase the stability of the sgRNA molecules and eliminate the type I interferon response seen with IVT gRNAs.18,33,34

Chemical modification of sgRNA and Cas9 delivered as mRNA produced efficient allelic disruption and integration rates in human CD34+ mobilized peripheral blood hematopoietic stem and progenitor cells.
Initial experiments compared the different endonuclease delivery methods for the ability to induce allelic disruption in the absence of a donor template. In this experiment, Cas9 mRNA vastly outperformed Cas9 RNP at the BTK intron 1 site (Fig. 5b), which is an observation we have not found true for any other target sites. Chemically modified sgRNA coelectroporated with Cas9 mRNA was by far the most effective combination of reagents, producing nearly 80% allelic disruption (Fig. 5b). The modified sgRNA used with recombinant Cas9 in RNP was less effective for allelic disruption at the intron 1 site, and the IVT sgRNA and Cas9 mRNA gave the lowest activity.
When a donor template was introduced to the cells via AAV6 transduction postelectroporation, successful integration events could be identified. The combination of chemically modified sgRNA with Cas9 mRNA also led to the highest frequencies (∼20%) of targeted integration (Fig. 5c). The levels of targeted donor integration with IVT sgRNA and Cas9 mRNA and with chemically modified sgRNA and Cas9 protein paralleled the activity of these nuclease combinations for gene disruption in the absence of the donor. Higher AAV6 vector multiplicities of infection generated higher rates of editing, however, also reduced the viability and expansion of the treated cells, suggesting cytotoxicity at higher vector doses (data not shown).
While delivering the Cas9 as mRNA with the sgRNA to intron 1 led to efficient editing of peripheral blood stem cells (PBSCs), the inefficiency of Cas9 delivered as RNP at this site prompted a search for additional target sites. Seven additional sgRNAs were assessed for their abilities to induce allelic disruption in BTK intron 1 or exon 2 in primary human CD34+ HSPCs, using RNP made with IVT sgRNA and Cas9 protein (Supplementary Fig. S3). This search identified a prime candidate sgRNA targeting BTK exon 2 that targets 7 bp after the BTK translational start codon, with high in silico predicted nuclease specificity.
Donor integration into BTK exon 2 maintains similar BTK expression levels with improved integration into human PBSCs with RNP
Plasmid donors were developed with homology arms for the BTK exon 2 site, following the same principles as for the intron 1 donors (Fig. 6a). Three variants were evaluated for expression levels generated from a BTK donor integrated at the BTK exon 2 target site: a base donor with the codon-optimized BTK cDNA, a BTK donor with I18t, and a donor containing both the WPRE and the I18u fragment. The codon optimization of the cDNA in the exon 2 donor disrupts the sgRNA binding site, and so, further PAM modification was not necessary. Each of these donor plasmids was delivered to BTK-deficient K562 cells via electroporation along with the exon 2 targeting sgRNA/Cas9 expression plasmid.
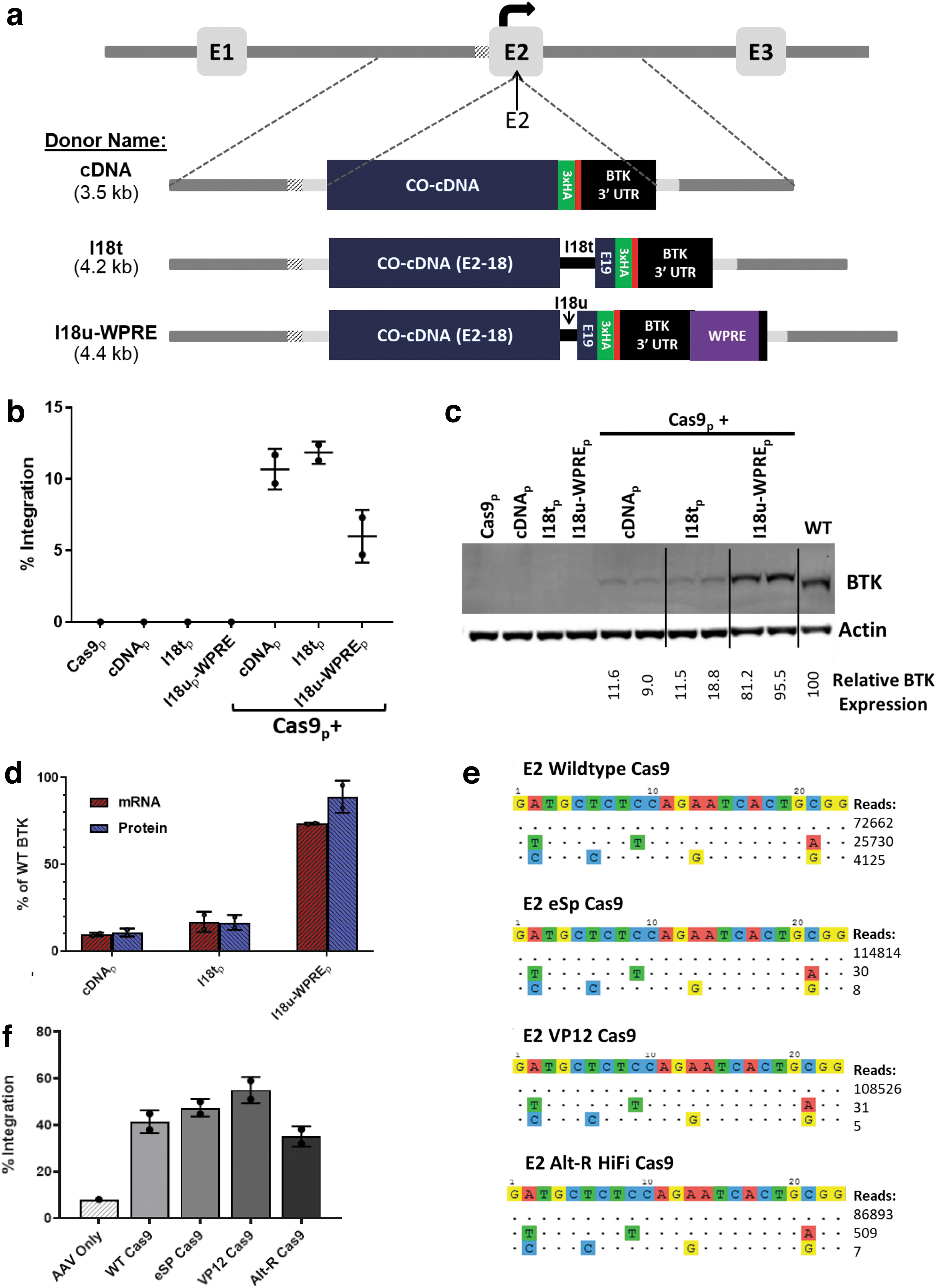
Targeted integration of a corrective cDNA donor into the BTK exon 2 target site led to efficient integration and expression of the transgene.
The base cDNA and I18t donors each generated over 10% BTK integration, while the I18u-WPRE donor integrated in 6% of cells (Fig. 6b). Probing protein lysates from these cells for BTK expression yielded a pattern similar to the pattern seen with the intron 1 reagents (Fig. 6c). The base donor generated much lower levels of BTK expression than wild-type K562 cells. Addition of the truncated intron 18 variant yielded a slight increase in BTK expression, while the microintron 18 fragment and WPRE together provided a large boost to expression, almost to wild-type BTK levels. The RNA expression levels by the BTK transgene very closely mirrored the quantified BTK protein levels from the immunoblot (Fig. 6d).
The exon 2 sgRNA was analyzed for its off-target nuclease activity in the K562 cell line using GUIDE-seq. 24 Two off-target loci yielded substantial cleavage events: intron 5 of the thrombospondin type 1 domain containing four genes and intron 21 of the low-density lipoprotein receptor-related protein 5 with 26.0% and 4.2% of the total cleavage events, respectively (Fig. 6e). To assess whether the off-target activity could be reduced, the exon 2 sgRNA was tested with three engineered Cas9 variants (eSP, VP12, and Alt-R HiFi) reported to have higher fidelity than the wild-type Cas9.19–21 All three variants nearly eliminated off-target activity, with 99.97%, 99.97%, or 99.41% on-target activity instead of only 70.88% with the wild-type protein (Fig. 6e).
These Cas9 variants were then tested with the exon 2 sgRNA for the ability to achieve targeted integration in human CD34+ PBSCs. The exon 2 sgRNA and each of the Cas9 variants were electroporated as RNP, while the exon 2 WPRE-I18u donor template was transduced as an AAV6 vector. All three Cas9 variants led to integration levels comparable with the wild-type Cas9 protein (Fig. 6f). Together, the specificity and activity data support the function of all three high-fidelity Cas9 variants for use with the sgRNA to exon 2 and identify this site with the I18u-WPRE donor as one achieving clinically relevant levels of integration and BTK expression.
Discussion
XLA is a promising candidate for gene therapy via targeted gene insertion for multiple reasons. First, the current standard of care has room for improvement, both in efficacy and duration. The only cure currently available, allogeneic HSC transplantation is rarely performed for XLA patients due to the associated risks of graft-versus-host disease, graft rejection, and transplant-related morbidities that generally outweigh the benefits of the treatment.
Autologous gene therapy offers similar potential benefits as allogeneic transplant with potentially far lower risks. Lentiviral vectors can be used to deliver a functional copy of BTK, although the drawbacks of nonphysiological regulation of the transgene and the added risk of IO may be untenable for treatment of a relatively mild disease. We propose that gene editing is the safest route to provide an enduring therapy for XLA.
Another aspect of XLA that makes it a good candidate for gene therapy is the strong selective advantage that B cells have with the functional BTK protein. In a murine model of the disease, low quantities of healthy HSPCs engrafted alongside BTK-deficient HSPCs were able to produce a disproportionally large percentage of mature B cells. 35 As few as 0.5% BTK-intact HSCs (out of 5 × 106 total cells) produced splenic B cells at frequencies higher than 10% of levels found in a healthy mouse as well as antibody responses to vaccine challenge within the normal range. 35
If this trend applies as expected in XLA patients, who have a more severe B cell deficiency than this murine model, the edited cells may have an even stronger selective advantage such that even a small fraction of edited HSCs would be sufficient for clinical benefit. This trait will help overcome one of the main challenges for current HDR-based gene editing therapies: achieving sufficient rates of gene modification in long-term HSCs.
The work presented here demonstrates the potential for BTK gene editing to achieve a lasting therapy for XLA. Initial work focused on achieving targeted integration into intron 1 of the BTK gene. The sgRNA used for this site led to reasonable allelic disruption frequencies when delivered as a plasmid in cell lines as well as no detectable off-target cleavage by GUIDE-seq. Experiments in K562 cells yielded consistently high levels of integration at this site. However, levels of BTK RNA and protein in cells were only 10–20% of wild-type levels.
The initial hypothesis of the project was that site-specific integration of the corrective BTK donor template into its endogenous locus would lead to near physiological expression of the gene, which these data do not fully support. The reduction in expression prompted a search for mechanisms to improve transgenic BTK expression without further disrupting the regulation of the gene. Although it remains unclear how much BTK expression per cell is necessary to provide a clinical benefit to XLA patients, previous work suggested that optimal BTK signaling efficiency occurs near wild-type expression levels. 11
The two main donor modifications that we identified to achieve that goals were the additions of a terminal intron and the WPRE. The terminal intron of the BTK gene improved expression of the transgene while preserving its lineage specificity. While the terminal intron has been reported to have distinct functions, it may be interesting to explore whether the presence of additional BTK introns further boosts protein expression. The WPRE also boosted expression levels, although it may have altered the lineage specificity of expression, with the unexpected observation of expression from the BTK transgenes that included the WPRE in Jurkat T cells. Readdition of elements from the endogenous BTK locus, such as the BTK cDNA, 3′ UTR, or introns, may have less intrinsic risk than adding exogenous elements such as the WPRE. The specificity of expression from these BTK transgenes in gene-edited HSCs after transplantation and multilineage differentiation needs to be examined to determine if it is both beneficial for restoring B lymphocyte function and safe without skewing leukocyte differentiation or activity. Animal models of the therapy are likely the best method to assess the safety of the treatment.
Integration of the BTK donors into human PBSCs also presented some unexpected difficulties. When the nuclease was delivered as IVT sgRNA with Cas9 protein, the intron 1 donor integrated at much lower than expected levels. Transitioning to a chemically synthesized (but unmodified) guide made no difference for gene editing frequencies. However, adding 2′-O-methyl and 3′ phosphorothioate internucleotide linkages on the terminal residues to stabilize the sgRNA tripled integration levels of the donor template. 18
The intron 1 site also had the unusual property that using Cas9 mRNA instead of recombinant Cas9 protein led to a large increase in BTK donor integration frequencies. We have not previously observed this strong preference for Cas9 mRNA over Cas9 RNP with any other target site. With these two changes to the delivery scheme for the intron 1 site (Cas9 mRNA and synthetic sgRNA with modified bases), integration levels of up to 30% were achieved. This integration frequency should be well above the threshold for clinical efficacy of gene editing for XLA. However, clinical-grade mRNA may be more difficult than recombinant Cas9 protein to procure in the quantity and quality necessary for use in clinical-scale editing. This concern spawned a search for alternative target sites near the 5′ end of the BTK locus and yielded a highly efficacious sgRNA targeting exon 2 of BTK.
The exon 2 sgRNA led to similar levels of editing in cell lines as the intron 1 sgRNA. However, unlike the intron 1 guide, the exon 2 guide had substantial off-target activity. Three recently developed high-fidelity Cas9 variants were tested for their abilities to reduce the off-target activity of the exon 2 sgRNA while maintaining the on-target activity. All three variants succeeded at nearly eliminating off-target activity while retaining most, if not all, of the on-target integration efficacy. Any of these variants could be considered for translation of BTK gene editing toward the clinic.
Conclusion
The work here constitutes major progress toward a clinically relevant gene editing scheme for an enduring treatment for XLA. Editing the BTK locus came with unanticipated hurdles that required the development of novel modifications to the BTK donor template as well as fine tuning of the reagent delivery scheme. The addition of the BTK terminal intron to the donor template led to a significant increase in BTK expression. We expect terminal intron addition to cDNA cassettes for improved expression to also be applicable and beneficial for other gene editing-based therapies that are in development. The WPRE provided an even larger expression boost, but it may have led to nonphysiological regulation of the BTK gene. To fully determine whether this expression we observed in T lymphoid lineages with BTK donors containing the WPRE is problematic, further studies in murine models of XLA will be performed. This work is a step toward improving the lives of XLA patients and provides insights that may facilitate the development of therapies for other genetic disorders.
Footnotes
Authors' Contributions
D.H.G., C.Y.K., and D.B.K. conceived of the course of study described here. Individual experiments were designed by D.H.G., I.V., C.Y.K., and D.B.K. D.H.G., I.V., J.L., J.S., A.K., and A.A. carried out the experiments, collected data, and analyzed the results. The article was initially drafted by D.H.G. and revised by D.H.G., C.Y.K., and D.B.K. We confirm that this article has been submitted solely to this journal and is not published, in press, or submitted elsewhere. All coauthors have reviewed and approved of the article before submission.
Acknowledgments
We would like to thank the Broad Stem Cell Research Center Flow Cytometry Core at UCLA for their guidance and assistance with fluorescence activated cell sorting.
Author Disclosure Statement
The work described here was partially funded by a sponsored research agreement with Orchard Therapeutics.
Funding Information
We would like to thank our funding sources: The Broad Foundation Innovative Pilot Stem Cell Research Project Award, The Immunology, Inflammation, Infection, and Transplantation Clinical and Translational Science Institute’s Discovery Award/David Geffen School of Medicine Seed Grant Program, Orchard Therapeutics, Ruth L. Kirschstein National Research Service Award GM007185 (DHG), Ruth L. Kirschstein National Research Service Award AI060567 (DHG), and the Institutional Research and Academic Career Development Award New York Consortium for the Advancement of Postdoctoral Scholars National Institutes of Health Award Number K12-GM102778 (DHG).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.