
Abstract
Adenine base editors (ABEs) can correct gene mutations without creating double-strand breaks. However, in recent reports, these editors showed guide-independent RNA off-target activities. This work describes our development of a delivery method to minimize ABEs' RNA off-target activity. After discovering a RNA off-target hot spot for sensitive detection of RNA off-target activities, we found that delivering ribonucleoproteins (RNPs) by electroporation generated undetectable non-specific RNA editing, but on-target base editing activity was also relatively low. We then explored a lentivirus capsid-based delivery strategy to deliver ABE. We used aptamer/aptamer-binding protein (ABP) interactions to package ABE RNPs into lentiviral capsids. Capsid RNPs were delivered to human cells for highly efficient guided base editing. Importantly, RNA off-target activities from the capsid RNPs were undetectable. Our new lentiviral capsid-based ABE RNP delivery method with minimal RNA off-target activities makes ABE one step closer to possible therapeutic applications.
Introduction
Fusing deaminases to nuclease-deficient type II CRISPR-Cas9 creates cytosine and adenine base editors (ABEs) that can edit genomic DNA without double-stranded DNA cleavage.1–3 Base editing generates precise point mutations in genomic DNA without generating double-strand breaks, requiring a DNA donor template or relying on cellular homology-directed repair. Thus, it has great potential as a technique to develop gene therapy for genetic diseases caused by transition mutations, which account for 61% of disease-causing point mutations. 4 ABEs have been used in many in vitro and in vivo studies. For example, ABE was used to correct a mutation in TERT promoter to inhibit brain tumor growth, 5 to correct nonsense mutations causing cystic fibrosis, 6 and to make multiplex precise base editing in cynomolgus monkeys. 7
However, in recent reports, ABEs showed guide-independent RNA off-target activities8,9 but minimal guide-independent DNA off-target activities.10–12 ABEs' guide-independent RNA off-target activities pose challenges to their potential clinical applications. Methods for reducing ABEs' RNA off-target activities are therefore needed to improve their safety.
One strategy is to engineer ABE variants with reduced RNA off-target activities.8,9,13–15 However, these ABE variants did not totally eliminate RNA off-target activities. Shortening the duration of activity of Cas9 via delivering Cas9 ribonucleoproteins (RNPs) can reduce off-target activities. 16 Similarly, delivering base editor RNPs can reduce guide-dependent DNA off-target activities,17,18 although its effects on reducing RNA off-target activities is unknown. Delivering ABE messenger RNA (mRNA) was shown to reduce RNA off-target activities greatly compared to delivering plasmid DNA, although it did not totally eliminate RNA off-target activities. 15 ABE protein purified from bacteria has been used in ABE DNA off-target profiling.19,20 Its application in base editing has not been reported.
We are interested in finding an ABE delivery method with short duration and minimal RNA off-target activities. With Cas9 and base editors, effector protein expression levels and expression duration contribute to specificity.16,17,21 Thus, ABE delivery methods featuring transient cargo expression and controllable amounts of cargo would be expected to result in reduced RNA off-target activities.
Recently, we used lentiviral (LV) capsids to deliver Cas9 mRNAs 22 or Cas9 RNPs23–25 for transient expression and efficient genome editing. Our strategy depends on the specific interaction between RNA aptamers and aptamer-binding proteins (ABPs) to package RNA/RNP into the LV capsids, in the absence of LV packaging signals or long terminal repeats.
ABP Com is a RNA-binding protein of 62 aa residues from the bacteriophage Mu that specifically binds to a short RNA aptamer (named as com with core sequence GAAUGCCUGCGAGCAUCC) observed in mom RNA.26,27 For packaging and delivering Cas9 RNPs, we fused ABP Com with nucleocapsid (NC) protein of the LV capsid precursor, and added aptamer com in the single-guide RNA (sgRNA). 23 Com–com interaction has been characterized before26,27 and used for CRISPR-based gene regulation. 28 The specific ABP–aptamer interaction efficiently packaged Cas9 mRNA or Cas9 RNP into the LV capsids.22,23 Highly efficient gene editing (>80% insertion or deletion rates) is achieved with LV capsid delivery methods.
Here, we tested whether ABE RNPs can be delivered by LV capsids to achieve high on-target base editing and negligible RNA off-target activities. LV capsid-delivered ABE RNPs showed high guided DNA base editing activities but undetectable guide-independent RNA off-target activities.
Methods
Plasmids
pMD2.G (Addgene #12259), pCMV_ABEmax (Addgene #112095) 29 and psPAX2-D64V (Addgene #63586) 30 were purchased from Addgene (Watertown, MA). The plasmid for expressing ABE7.10 in Escherichia coli has been described previously. 19 Other plasmids were generated by this group, as shown in Supplementary Table S1 or as described recently.22,23 Plasmids will be made available through Addgene (IDs: 132554, 132560, and 136270) or on request. Gene synthesis was done by GenScript, Inc. All constructs generated were confirmed by Sanger sequencing. Sequence information for primers and oligoes are listed in Supplementary Table S2. ABE target sequences and the oligos used for making the sgRNA expression constructs are listed in Supplementary Table S3.
ABE 7.10 expression and purification
The SNU-ABE plasmid, which encodes codon optimized ABE 7.10 linked to an N-terminal His tag, 19 was first transformed into BL21-star (DE3) competent cells, which were then plated on a Luria-Bertani (LB) agar plate containing 50 μg/mL kanamycin. After incubation overnight at 37°C, a single colony was selected and grown overnight at 37°C (pre-culture) in LB broth containing 50 μg/mL kanamycin and 10 μM ZnCl2 to maintain ABE catalytic activity. Following this pre-culture, part of the inoculant was transferred to several 400 mL LB media in a 1 L flask for large culture (up to 6 L), and the resulting culture was incubated at 37°C with shaking at 250 rpm until the absorbance A600 = ∼0.5–0.70. Next, the culture was put on ice for about 1 h. To induce ABE protein expression, 1 mM isopropyl β-D-1-thiogalactopyranoside (GoldBio, St. Louis, MO) was added, and the culture was incubated at 18°C for 14–16 h with 250 rpm shaking.
The later steps in the purification procedure were all carried out at 0–4°C. Prior to cell lysis, the cells were harvested by centrifugation at 5,000 g for 10 min, after which they were re-suspended in 8 mL lysis buffer per 400 mL inoculants (50 mM sodium phosphate, 500 mM 1% Triton X-100, 20% glycerol, 1 mM phenylmethylsulfonyl fluoride, 1 mg/mL lysozyme from chicken egg white, 10 μM ZnCl2, pH 8.0; all Sigma–Aldrich. St. Louis, MO). For lysis, cells were frozen in liquid nitrogen and thawed at 37°C a total of three times. For further lysis, cells were sonicated (3 min total, 5 s on, 10 s off), after which they were centrifuged at 13,000 g to clear the lysate. The supernatant was mixed with 10 mL Ni-NTA agarose beads (Qiagen, Valencia, CA); the resin–lysate mixture was gently rotated for 1 h and then loaded onto a column. The column was washed three times each with 50 mL nickel wash buffer (50 mM sodium phosphate [Sigma–Aldrich], 150 mM NaCl [Sigma–Aldrich], 35 mM imidazole [Sigma–Aldrich], 1 mM DTT [GoldBio], 10 μM ZnCl2 [Sigma–Aldrich], pH 8.0), and then the proteins were eluted with 20 mL nickel elution buffer (50 mM sodium phosphate, 150 mM NaCl, 250 mM imidazole, 20% glycerol, 1 mM DTT, 10 μM ZnCl2, pH 8.0). The eluted proteins were further purified with 5 mL heparin Sepharose beads (GE Healthcare, Chicago, IL) in another column. The column was washed with 50 mL heparin wash buffer (50 mM sodium phosphate, 150 mM NaCl, 1 mM DTT, 10 μM ZnCl2, pH 8.0) three times, and proteins were eluted with 20 mL heparin elution buffer (50 mM sodium phosphate, 750 mM NaCl, 20% glycerol, 1 mM DTT, 10 μM ZnCl2, pH 8.0). Finally, the eluted proteins were concentrated and the buffer changed to ABE storage buffer (200 mM NaCl, 20 mM HEPES, 1 mM DTT, 40% glycerol, pH 7.5) by centrifugation through an Amicon Ultra-4 column with a 100,000 kDa cutoff (Millipore, Burlington, MA) at 6,000 g.
ABE- and Endo V-mediated in vitro digestion of amplified target site
The region spanning the ABE site 1 (Hek2) was amplified using polymerase chain reaction (PCR; chr5:+87944480-87944802) with primers HEK2-F and HEK2-R. The resulting amplicon (2 μg) was then incubated with 4 μg ABE 7.10 protein and 3 μg sgRNA (targeting ABE site 1) in 200 μL ABE reaction buffer (50 mM Tris-HCl [Sigma–Aldrich], 25 mM KCl [Sigma–Aldrich], 2.5 mM MgSO4 [Sigma–Aldrich], 0.1 mM ethylenediaminetetraacetic acid [EDTA; Sigma–Aldrich], 2 mM DTT [GoldBio], 10 mM ZnCl2 [Sigma–Aldrich], 20% glycerol) at 37°C for 1–2 h. Following the reaction, ABE protein and sgRNA were removed by incubation with 80 μg Proteinase K and 400 μg RNase A (both from Qiagen), respectively, for 10 min. The amplicons were purified using a PCR purification kit (MGmed, Seoul, Korea). The purified amplicons (1 μg) were incubated with 10 IU of Endo V enzyme (NEB, Ipswich, MA) for 1 h. Next, the mixture was incubated with 80 μg Proteinase K, and again purified with a PCR purification kit (MGmed). Finally, the DNA fragments were imaged following electrophoresis on a 2% agarose gel.
RNP electroporation
CRISPR RNA for ABE site 1 (rGrArArCrArCrArArArGrCrArUrArGrArCrUrGrCrGrUrUrUrUrA rGrArGrCrUrArUrGrCrU) was synthesized by IDT, Inc. (Coralville, IA). Alt-R®CRISPR-Cas9 tracrRNA, Alt-R® CRISPR-Cas9 Negative Control crRNA, Alt-R® Cas9 Electroporation Enhancer, and Nuclease Free Duplex Buffer were purchased from IDT, Inc. RNP reconstitution and electroporation were performed following the manufacturer's instructions. A total of 2 × 105 HEK293T cells were used for each electroporation with the Amaxa Nucleofector system (Lonza, Basel, Switzerland). The cells were re-suspended in 100 μL nucleofection buffer from the Cell Line Nucleofector™ Kit V (catalog #VCA-1003; Lonza) and placed in the electroporation cuvette. Then, 1 μL Alt-R® Cas9 Electroporation Enhancer and 5 μL re-constituted ABE RNPs were added to the cells in the cuvette. Finally, the cells were given an electric shock with protocol Q-001. The cells were removed from the cuvette and cultured in growth medium for 24 h before analysis.
LV capsid RNP production
LV capsids packaged with ABE RNPs were produced by a three-plasmid transfection procedure. Briefly, 13 million HEK293T cells were cultured in 15 cm dish with 15 mL Opti-MEM. Then, 16 μg ABP-modified packaging plasmid pspAX2-D64V-NC-ABP (ABP could be MCP [
Concentrating ABE RNP-laden VLPs
The supernatant containing ABE RNP-laden VLPs was concentrated with the KrosFlo® Research 2i Tangential Flow Filtration System (Cat. No. SYR2-U20; Spectrum Laboratories, Inc., Piscataway, NJ) using the concentration-diafiltration-concentration mode. Briefly, 150–300 mL supernatant was first concentrated to about 50 mL, diafiltrated with 500–1,000 mL phosphate-buffered saline, and finally concentrated to about 8 mL. The hollow-fiber filter modules were made from modified polyethersulfone, with a molecular weight cutoff of 500 kDa. The flow rate and the pressure limit were, respectively, 80 mL/min and 8 psi for the filter module D02-E500-05-N and 10 mL/min and 5 psi for the filter module C02-E500-05-N. Capsid RNPs were also concentrated by ultracentrifugation, as described previously. 22
VLP quantification
Concentration of VLPs was determined by p24 (LV capsid protein CA)-based enzyme-linked immunosorbent assay (QuickTiter™ Lentivirus Titer Kit, catalog number VPK-107; Cell Biolabs, San Diego, CA). When unconcentrated samples were assayed, the VLPs were precipitated according to the manufacturer's instructions so that the soluble p24 protein was not detected.
Western blotting analysis of capsid and Cas9 proteins in ABE RNP VLPs
A published procedure was used to treat 200 ng p24 of VLPs transiently with 0.5% Triton X-100. 32 Briefly, VLPs were centrifuged with a Sorvall T-890 rotor (2 h at 120,000 g) through step gradients containing a 1 mL layer of 10% sucrose in STE (100 mM NaCl, 50 mM Tris/HCl, pH 7.5, 1 mM EDTA) with or without 0.5% Triton X-100, and a cushion of 2 mL 20% sucrose in STE solution. The pelleted VLP particles were directly lysed in 100 μL of 1 × Laemmli sample buffer for Western blotting or for purifying RNA for reverse transcription quantitative (RT-qPCR) analysis.
The proteins in each sample were separated on sodium dodecyl sulfate polyacrylamide gels and analyzed by Western blotting. The antibodies used include mouse monoclonal anti-SpCas9 antibody (CRISPR-Cas9 Monoclonal Antibody 7A9-3A3, catalog # MA1-201, 1:1,000; Thermo Fisher Scientific, Waltham, MA), and p24 mouse monoclonal antibody for capsid protein (catalog # 310810, 1:1,000; Cell Biolabs). Horseradish peroxidase–conjugated anti-mouse IgG (H + L; catalog # 31430, 1:5,000; Thermo Fisher Scientific) and anti-Rabbit IgG (H + L; catalog # 31460, 1:5,000) secondary antibodies were used in Western blotting. SpCas9 RNP standards were GenCrispr NLS-Cas9-NLS Nuclease from GenScript (catalog # Z03389S). Chemiluminescent reagents (Pierce, Rockford, IL) were used to visualize the protein signals in the LAS-3000 system (Fujifilm, Tokyo, Japan). Densitometry (ImageJ software v49; National Institute of Health, Bethesda, MD) was used to quantify protein amounts.
RNA isolation and RT-qPCR analysis
A miRNeasy Mini Kit (catalog # 217004; Qiagen) was used to isolate RNA from concentrated capsids or cells. The QuantiTect Reverse Transcription Kit (Qiagen) was used to reverse transcribe the RNA to cDNA. For sgRNA reverse transcription, 0.6 μL random primers provided in the kit and 0.4 μL sgRNA-specific primer (Sp-sgRNA-R1, gcaccgactcggtgccactt, 20 μM) were used for reverse transcription. Then, guide-specific forward primer ABE-g5-F (Supplementary Table S2) was used together with Sp-sgRNA-R1 in SybrGreen based RT-qPCR to detect sgRNA. Quantitative PCR was run on a QuantStudio 3 or ABI 7500 instrument.
VLP transduction
VLPs (in 10–300 ng p24 protein) were added to 2.5 × 104 cells grown on 24-well plates with 8 μg/mL polybrene. Unconcentrated supernatant of VLPs was diluted with fresh medium at a 1:1 ratio to transduce cells. The cells were incubated with the particle containing medium for 12–24 h, after which the medium was replaced with normal medium.
Examination of ABE protein degradation in cells
HEK293T cells (2 × 104) were transduced with 100 ng p24 of VLPs containing ABE RNPs with or without aptamer. Twelve hours after transduction, the cells were maintained in Dulbecco's modified Eagle's medium with 0.5% fetal bovine serum to limit cell division. The medium was changed every 48 h. Cells were collected every 12 h after transduction to detect the presence of ABE protein by Western blotting using anti-SpCas9 (catalog # MA1-201; Thermo Fisher Scientific) and anti-β actin (A5441, 1:5,000; Sigma–Aldrich) antibodies. The relative expression of ABE was quantified by densitometry with ImageJ. The densitometry data were used to determine protein half-life using the two-phase decay method of GraphPad Prism v5.0 (GraphPad Software LLC, La Jolla, CA).
Next-generation sequencing and data analysis
The regions and primers used to amplify target DNA for next-generation sequencing (NGS) are listed in Supplementary Table S2. The proofreading HotStart® ReadyMix from KAPA Biosystems (Wilmington, MA) was used for PCR. The amplicons were sequenced by GeneWiz's Amplicon-EZ service. Usually, 50,000 reads/amplicon were obtained. Base editing was analyzed with the online software BE analyzer, 33 and CRISPRESSO2, 34 which gave similar results.
Statistical analysis
GraphPad Prism v5.0 was used for statistical analyses. t-Tests were used to compare the averages of two groups. Analysis of variance (ANOVA) was performed followed by Tukey post hoc tests to analyze data from more than two groups. Bonferroni post hoc tests were performed following ANOVA in cases of two factors. A p-value of <0.05 was regarded as statistically significant.
Results
Finding RNA hot spots for detecting ABE RNA off-target activities
The major goal of this study was to find an ABE delivery method with short activity duration and minimal RNA off-target activities, for which a sensitive RNA off-target detection method will be useful. Currently, high-depth RNA sequencing is used to detect ABE RNA off-target activities,8,9 which is time-consuming and expensive. Recently, it was found that the RNA motif CUACGAA was the most efficient ABE RNA off-target. 13 We analyzed human sequence database and found that the human USP38 gene contains a CTACGAA sequence in its coding region exon 9 (Fig. 1A). RT-PCR confirmed that this gene was expressed in HEK293T cells. We wondered whether the “CUACGAA” sequence of USP38 mRNA is an ABE RNA off-target hot spot.
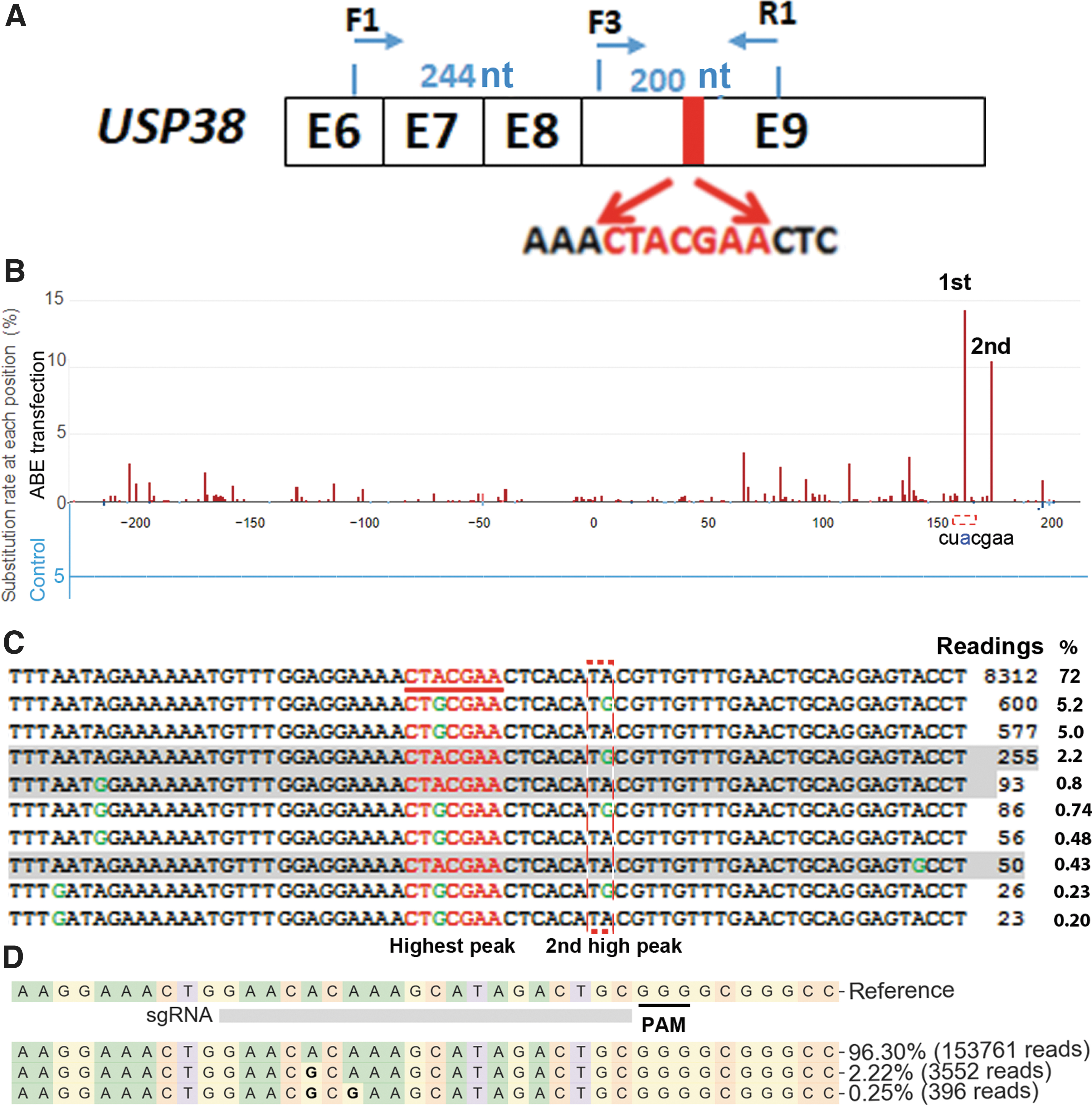
Searching for adenine base editor (ABE) RNA off-target hot spots.
We transfected HEK293T cells with plasmid DNA expressing Cas9 nickase (negative control) or plasmid DNA expressing ABE and sgRNA targeting ABE site 1.
2
We amplified 444 bp of the USP38 cDNA spanning the predicted hot spot (primers F1 and R1 in Fig. 1A) for targeted NGS. In cells transfected with ABE-expressing DNA, we observed the highest peak of “A” to “G” change at the predicted hot spot CU
These “A” to “G” changes must be the results of changes in mRNA, since NGS analysis of corresponding DNA amplified from genomic DNA of cells transfected with ABE and ABE site 1 sgRNA revealed an A to G change in <0.02% of alleles. The changes observed in USP38 cDNA were most likely the results of nonspecific RNA editing of adenosine (A) to inosine (I), which was recognized as Guanine (G) in reverse transcription and sequencing. The most frequently observed A to G changes all occurred in the UA motif, consistent with previous observations (Fig. 1C).8,9,13
Focusing on the A to G changes in the “CU
Only reads with CU(/T)
All reads were from one NGS sample.
NGS, next-generation sequencing.
ABE RNPs delivered by electroporation showed undetectable RNA off-target activities 24 h after delivery
Once we confirmed an ABE RNA off-target hot spot, we tested whether delivering ABE RNPs by electroporation showed reduced RNA off-target activity compared to DNA transfection. We prepared recombinant ABE RNPs, as we recently described, 19 and confirmed their activities in an in vitro assay (Supplementary Fig. S1). We delivered 20, 10, 5, 2.5, 1.25, and 0.625 μg of ABE RNPs (targeting ABE site 1) into 2 × 105 HEK293T cells by electroporation. We designed primers specific for DNA with base editing and verified that this qPCR assay yielded cycle threshold (Ct) values differing by ∼6 when comparing DNAs from nickase-transfected cells versus ABE-transfected cells, validating this approach. Twenty-four hours after treatment, qPCR detected on-target base editing in cells treated with 20 and 10 μg ABE RNPs but not in cells treated with lower amounts of ABE RNPs. We thus performed NGS to examine on-target base editing in cells treated with 20 and 10 μg ABE RNPs, and observed 2.10 ± 0.22% (n = 3) and 1.93 ± 0.53% (n = 3) on-target base editing, respectively (Fig. 1D). These were occurrences of target-specific base editing, since electroporation of ABE RNPs with a random sgRNA showed A to G changes at ABE site 1 in only 0.01% of samples.
We then examined RNA off-target activities at the USP38 hot spot. We observed no off-target RNA editing at the USP38 hot spot in any of the six samples, which was in sharp contrast to the high level (>15%) of RNA off-target editing with ABE plasmid DNA transfection (Fig. 1C and Table 1). The data indicate that ABE RNPs showed detectable on-target DNA editing but undetectable off-target RNA editing 24 hours after delivery.
Although delivering ABE RNPs by electroporation greatly reduced RNA off-target activities, relatively low on-target base editing (<5%) occurred after electroporation of 20 μg (∼100 pmol) ABE RNPs, possibly due to ABE's relatively large protein size (∼1,800 aa residues). It could be difficult to improve on-target base editing efficiency significantly simply by increasing the dosage. Thus, a more efficient ABE RNP delivery method is needed.
Developing ABE RNP-laden VLPs via packaging ABE RNPs in LV capsids
We recently showed that the aptamer–ABP interactions could be used to package Cas9 RNPs into LV capsids for efficient genome editing. 23 Here, we tested whether a similar strategy could be used for ABE RNP delivery. Considering the different sizes of the proteins in question (∼1,800 aa for ABE vs. 1,114 aa for SaCas9) and that the Cas9 proteins were from different species (Streptococcus pyogenes for ABE vs. Staphylococcus aureus for SaCas9) and had different sgRNA scaffolds, we decided to test three ways of sgRNA scaffold modification. First, we used an MS2 aptamer to replace both the Tetraloop and the ST2 loop because this modification worked in endonuclease null Cas9 (dCas9)-mediated DNA labeling (Fig. 2A). 35 Second, we used one copy of a com aptamer to replace the Tetraloop loop, as we recently described. 23 Third, we used one copy of com aptamer to replace the ST2 loop, since we recently found that this modification was the most efficient for packaging SpCas9. 24 One copy of the aptamer was tested, since we and others found that more than one copy greatly decreases RNA stability.22,28
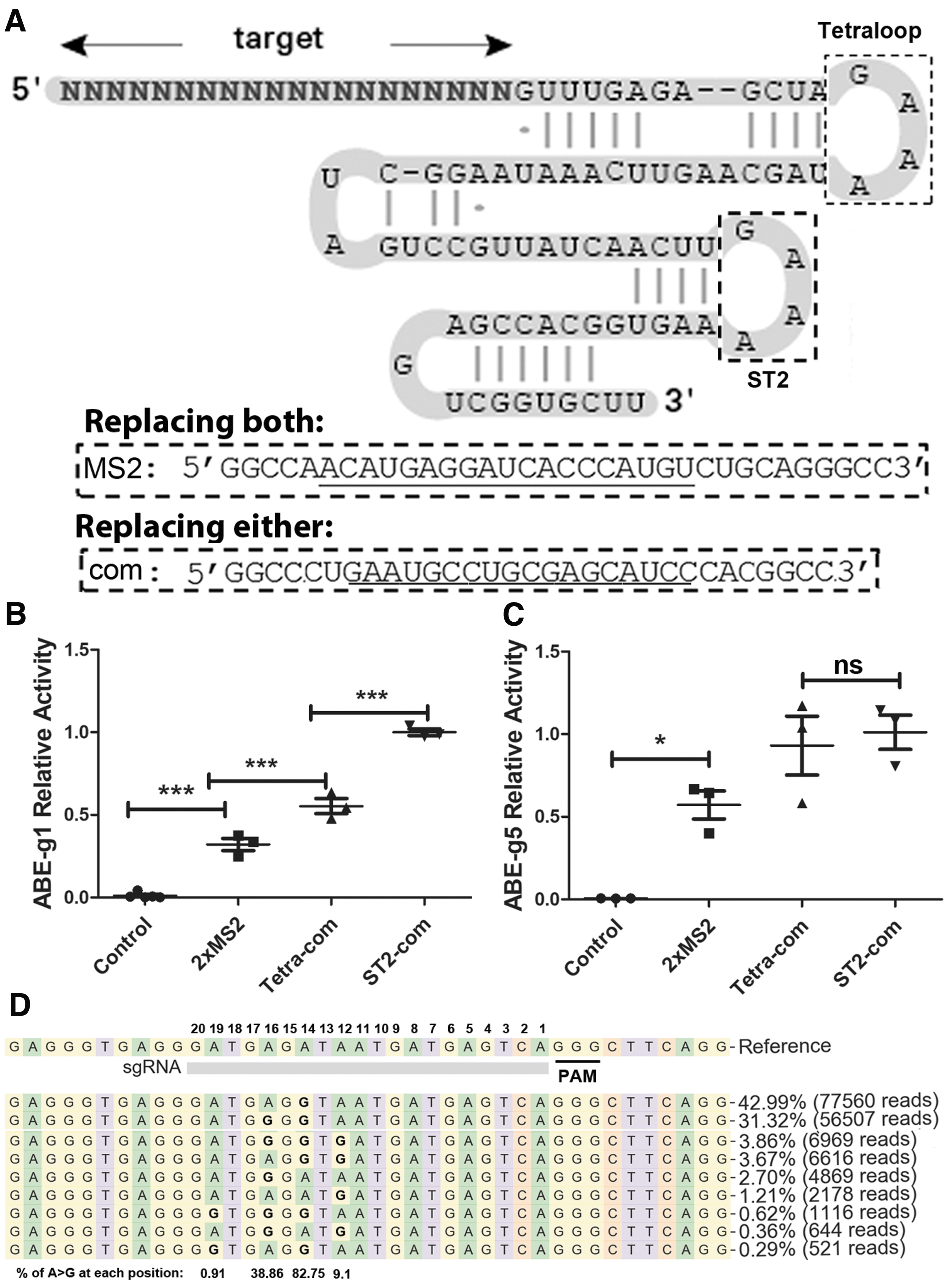
Modifying single-guide RNA (sgRNA) for ABE RNP package and delivery by lentiviral (LV) capsids.
We packaged ABE-RNP into LV capsids by co-transfecting three plasmids into HEK293T cells: the envelope plasmid pMD2.G expressing the VSV-G protein, the target plasmid co-expressing ABE and various target-specific aptamer-modified sgRNAs, and the packaging plasmids modified by the corresponding ABPs (pspAX2-D64V-NC-MS2 for MS2 modified sgRNA and pspAX2-D64V-NC-Com for com modified sgRNAs), as described recently.22–24 The supernatants containing capsid/ABE RNPs were used to transduce HEK293T cells. Then, we compared their base editing activities with qPCR.
sgRNA g1 and g5 were used to target ABE sites 1 and 5, respectively. These were the two sites previously shown to be successfully edited after transfecting the corresponding ABE-expressing plasmid DNA. 2 We then used qPCR to detect the base editing activities of capsid/ABE RNPs packaged with sgRNA containing 2 × MS2, Tetra-com, and ST2-com, respectively. We detected 20–160 times more edited products in capsid/ABE RNP-treated cells than in negative control cells (ABE-g5 RNP-treated cells as controls for ABE-g1 RNP-treated cells and vice versa) at ABE sites 1 and 5. All three types of ABE RNPs were functional (Fig. 2B and C).
For ABE sites 1 and 5, 2 × MS2 modification showed the least base editing activity. For ABE site 5, the activities of single-copy com modified sgRNAs showed similar activities at the Tetraloop and ST2 loop locations. However, for ABE site 1, ST2-com modified RNPs performed significantly better than Tetra-com modified RNPs (p < 0.0001). We thus used ST2-com modification of sgRNA for further experiments. The aptamer–ABP strategy was able to package and deliver functional ABE RNPs to human cells.
We further examined the base editing activity of the ABE RNP VLPs by NGS. When targeting ABE site 1 in 2.5 × 104 HEK293T cells, 200 ng p24 of capsid-ABE RNPs generated A to G editing in 31.85% alleles (Supplementary Fig. S2). When targeting ABE site 5 in 2.5 × 104 HEK293T cells, 108 ng p24 of capsid-ABE RNPs (non-concentrated supernatant) generated A to G editing in 87.5% of all alleles (Fig. 2D). In contrast, in cells treated with VLPs targeting ABE site 5, we observed A to G change in 0.02% of alleles at ABE site 1, and in cells treated with VLPs targeting ABE site 1, we observed A to G change in 0.01% of alleles at ABE site 5. These data show that the VLPs generated high-level site-specific base editing.
Aptamer-dependent assembling of ABE RNP-laden VLPs
We examined whether aptamer–ABP interaction was necessary for the RNPs to be packaged inside the capsids as designed. We compared ABE protein content in capsids with ABE-g5 RNP (unmodified g5 sgRNA) and ABE-g5ST2-com RNP (ST2-com modified g5 sgRNA). To eliminate possible ABE protein associated with vesicles or the particle membrane, we transiently treated the particles with 0.5% Triton X-100 buffer, 32 which removed membrane-associated SaCas9 protein in our previous study. 23 We confirmed that this procedure reduced capsid protein p24 by >100% (Fig. 3A).
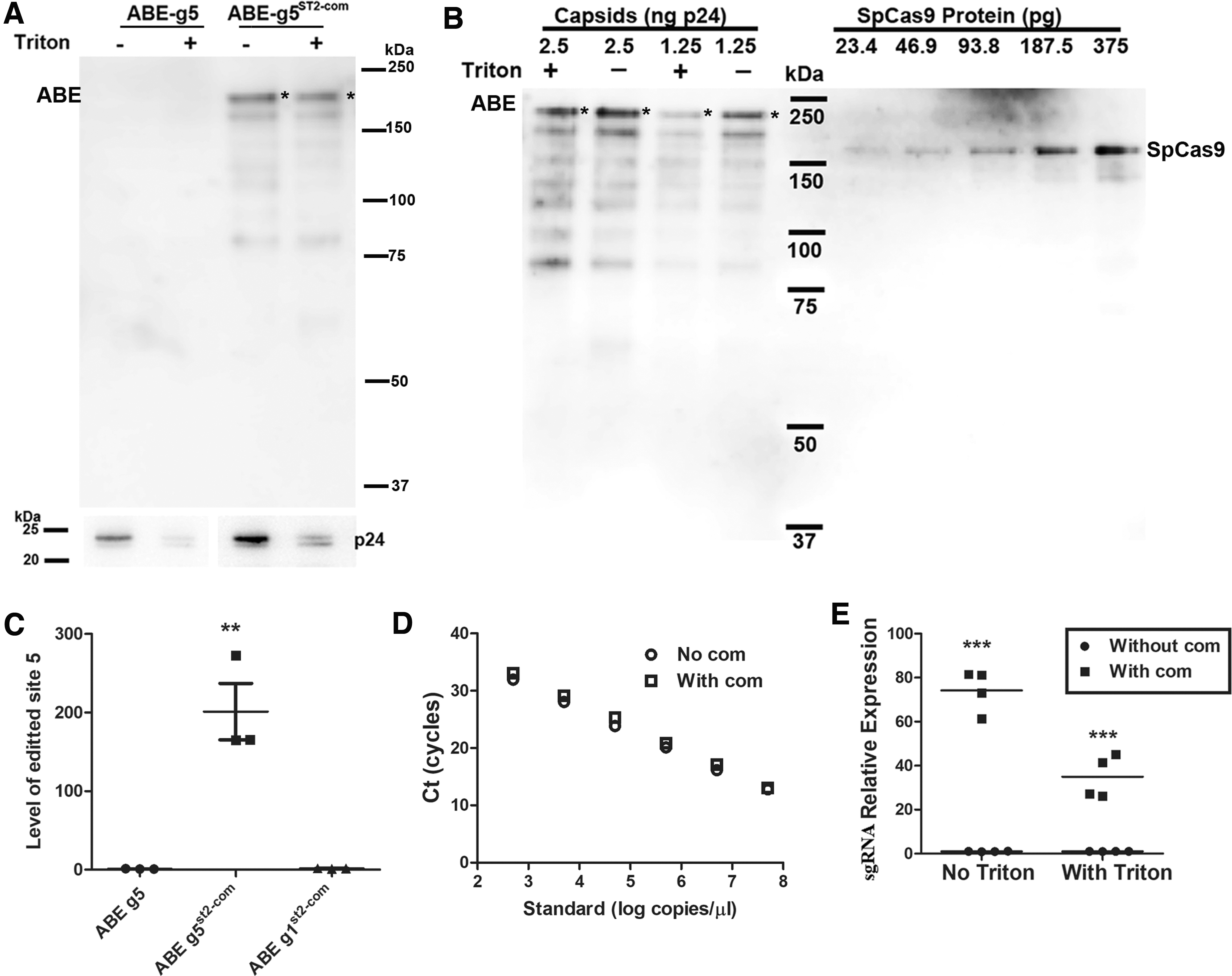
Aptamer–ABP interaction is necessary for functional ABE packaging in LV capsids.
We then examined ABE protein by Western blotting with SpCas9 antibody. ABE was only detected in capsids with ABE-g5ST2-com RNPs but not in capsids with ABE-g5 RNPs (Fig. 3A). In addition, transient 0.5% Triton X-100 treatment decreased ABE amounts by 30–50%. Compared to SpCas9 proteins of known concentration, the ABE amount in Triton-treated capsids was about 100 pg ABE/ng p24 (Fig. 3B, only considering the full-length ABE with an asterisk). Assuming 1.25 × 107 capsids per ng p24, the ABE molecule numbers per capsid were estimated at 30 molecules per capsid.
Consistent with the lack of ABE protein in ABE-g5 RNP capsids, qPCR failed to detect base editing activities in cells treated with capsids packaged with ABE-g5 (without st2-com) RNPs (Fig. 3C). The data showed that ABE association with the capsids and base editing activities were aptamer dependent.
We also compared sgRNA levels in the VLPs by RT-qPCR. We first performed qPCR using known concentrations of the respective plasmid DNA (with or without com in sgRNA) to confirm that the com aptamer did not affect qPCR detection (Fig. 3D). In equal amounts (300 ng p24) of VLPs treated with and without Triton, the levels of g5ST2-com sgRNA (with com) were 35.0 ± 4.8 (n = 4) and 74.2 ± 4.8 (n = 4) times those of g5 sgRNA (without com), respectively (Fig. 3E). The sgRNA qPCR data are consistent with our Western blotting data showing that com modification of sgRNA increased ABE levels in capsids and Triton X-100 treatment decreased it. Together, the data showed that packaging of ABE protein and sgRNA in the capsids and base editing activity of the VLPs all depended on com modification of sgRNA, confirming the role of ABP–aptamer interaction in packaging ABE RNPs.
VLPs enable transient expression of ABE RNPs in human cells
To determine the expression duration of ABE RNPs in human cells, we transduced ABE-g5ST2-com RNP-laden VLPs and ABE-g5 RNP-laden VLPs (each 100 ng p24/well) into HEK293T cells and measured ABE protein levels every 12 h. In RNP-treated but not control cells, Western blotting detected a band between 150 and 250 kDa (Fig. 4A), consistent with the expected size of ABE (204.7 kDa). In cells transduced with ABE-g5 RNP capsids, we observed a random fluctuation of low ABE levels (<25% of highest ABE-g5ST2-com RNP level at all time points). In cells transduced with ABE-g5ST2-com RNP capsids, ABE levels were highest during the first 24 h post transduction and reduced slightly at 24–48 h post transduction. At 48–72 h post transduction, ABE levels dropped to ∼25% of levels at 12 h post transduction, similar to levels in cells treated with ABE-g5 RNPs. At ∼60 h post transduction, ABE levels were half of those at 12 h post transduction (Fig. 4A and B).
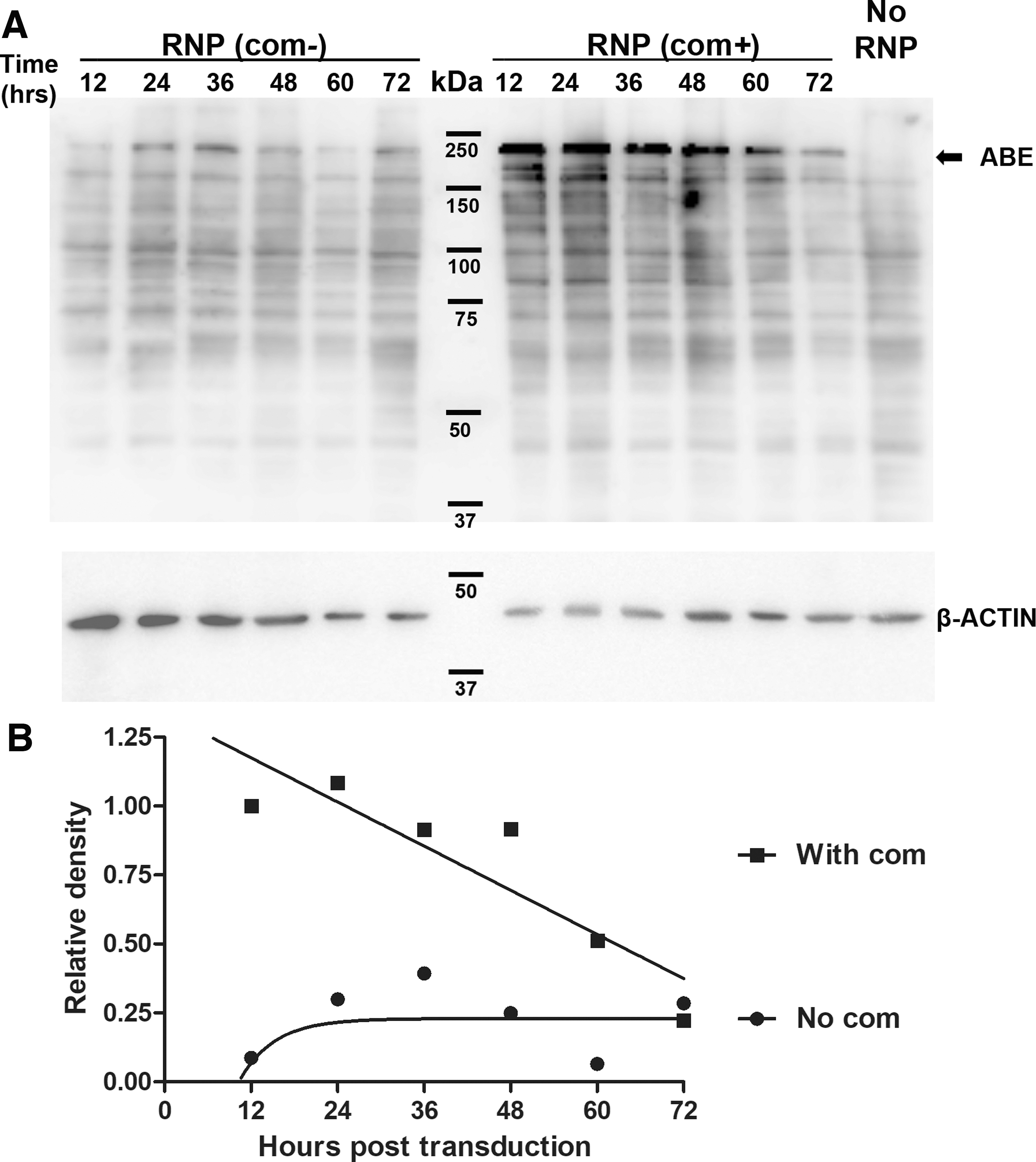
Western blotting analysis of ABE levels after transducing HEK293T cells.
In the experiment examining ABE in VLPs (Fig. 3A), we did not detect ABE in ABE-g5 RNP VLPs. In that experiment, ABE-g5 RNP VLPs were subjected to an ultracentrifugation in a buffer without Triton X-100, and VLPs used to transduce cells were not centrifuged. We reasoned that the low background ABE in cells transduced with ABE-g5 RNP VLPs were likely the ABE in the capsid preparation. This was concentrated by the tangential low filtration system but not packaged in the capsids, and thus could be removed by ultracentrifugation. The data confirmed the short-term expression of ABE RNPs delivered by VLPs.
ABE RNP-laden VLPs showed undetectable guide-independent RNA off-target activities
We then examined whether ABE RNPs delivered by LV capsids generated detectable RNA off-target activities. We targeted ABE site 1 by ABE RNP-laden VLPs and plasmid DNA transfection. We first determined the conditions for the two delivery methods giving similar on-target base editing efficiencies. We examined on-target and off-target activities 24 h after treatment, since that was the time point with the highest ABE level after VLP treatment. qPCR analysis of gDNA 24 h after treatment revealed that transfection of 250 ng plasmid DNA showed similar gene editing activity on ABE site 1 as transducing 100 ng p24 of capsid RNPs. We then performed NGS on ABE site 1 genomic DNA and USP38 cDNA (amplified with F3 and R1 in Fig. 1A). ABE site 1 DNA had a slightly higher on-target A to G base editing rate in capsid RNP-transduced cells (14.5%) than in plasmid DNA-transfected cells (9.2%; Table 2).
p < 0.05.
We then analyzed RNA off-targets around the USP38 hot spot. Since in previous experiments we observed a second peak near the predicted hot spot (peak 2 in Fig. 1B), we examined the percentages of A to G changes at both peaks. In VLP-treated cells, we observed A to G change rates similar to negative control cells at both peaks, whereas in plasmid DNA-transfected cells, significantly higher A to G change rates occurred at both peaks compared to VLP-treated cells (Table 2). In this experiment, DNA transfection resulted in ∼20 times lower RNA off-target rates than the DNA transfection experiment described in Figure 1 (0.667% vs. ∼15% for the hot spot). The lower level of RNA off-target activities in this experiment could have been caused by two non-exclusive mechanisms: (1) less DNA was transfected (250 ng vs. 500 ng), and (2) RNA off-target activity was detected 24 h rather than 48 h after transfection. Nevertheless, delivering ABE RNPs by LV capsids caused undetectable RNA off-targets, although the on-target DNA base editing level was 56% higher than in cells treated with DNA transfection.
We examined RNP off-target activities 24 h after VLP delivery because our ABE RNP expression duration data showed that ABE RNPs were highest 24 h after transduction (Fig. 4A). We also examined RNA off-targets 48 h after VLP delivery and observed no RNA off-target activities at the hot spot (Supplementary Fig. S3). Since ABE protein levels decreased quickly after this time point, it is unlikely that further RNA off-target activities could be detected later. Thus, ABE RNPs delivered by LV capsids showed RNA off-target activity below our detection limit.
Discussion
This work attempted to find an ABE delivery method with short activity duration, high base editing efficiency, and minimal RNA off-target activity. Two of our observations may help to resolve the safety concerns caused by ABE's RNA off-target activities, especially for in vivo applications. First, delivering ABE RNPs generated detectable on-target DNA base editing with undetectable RNA off-target activities. Second, we developed novel ABE RNP-laden VLPs with high on-target DNA base editing efficiency and undetectable RNA off-target activity.
RNPs have been used in genome editing and cytosine base editing with improved specificity.16,17 We tested delivering ABE RNPs by electroporation and observed relative low base editing activity (<5%) when using ABE RNP amounts common to Cas9 RNP electroporation protocols, although using more ABE RNPs in electroporation may improve base editing activity. We then developed ABE RNP-laden VLPs using our recently developed Cas9 LV capsid delivery system.23,24 We packaged ∼30 ABE RNP molecules into each capsid particle. When targeting ABE site 1 in HEK293T cells, ABE RNP electroporation resulted in <5% base editing efficiency at 5 pg/cell (10 μg RNPs for 2 × 105 cells), whereas ABE RNP VLP transduction resulted in >30% base editing efficiency at 0.8 pg/cell (∼20 ng RNPs for 2.5 × 104 cells). When targeting the ABE g5 site, we obtained >85% base editing efficiency at the dose of 0.43 pg/cell. Thus, ABE RNP-laden VLPs resulted in much more efficient base editing, although much less ABE protein was used. Our novel ABE RNP-laden VLP is the first ABE RNP delivery vehicle demonstrating high base editing activity and low RNA off-target activity.
VLPs have been successfully used to package and deliver Cas9 nuclease via two different strategies: (1) fusing Cas9 with a viral capsid precursor,36–38 and (2) using interactions between RNA aptamer and ABP.23,24 Fusing Cas9 with viral capsid precursor greatly impaired capsid assembly. Even after rescuing capsid assembly with unmodified capsid precursors, the capsid assembly efficiency was 20–30% of that of normal capsid assembly. The inefficient capsid assembly makes production of Cas9 RNP-laden VLPs challenging. We used the aptamer–ABP interaction to package ABE RNPs into LV capsids and typically obtained ∼100 ng p24 capsid/mL supernatant, in the same range as making normal LV particles.
In addition to the high capsid assembly efficiency and base editing efficiency (>80% editing efficiency with unconcentrated VLPs), we observed no RNA off-target activities 24 h after VLP delivery. We cannot completely rule out RNA off-target generation before our detection. However, previously we found that the earliest time to observe gene editing activity after delivering VLPs was 16 h post transduction. 23 Since escaping from the endosome system is a similar process to VLPs entering recipient cells, a comparable time should be needed for ABE RNPs to become functional after delivery. RNA off-targets, if any, could have been generated 16–24 h after RNP delivery. This short time window may greatly reduce the chances of generating enough erroneous proteins to be harmful to the cells. Delivering ABE mRNA has reduced but still detectable RNA off-target activities. 15 Thus, delivering ABE RNP by VLPs is safer due to the undetectable RNA off-target activities.
Our data show that VLP is an efficient ABE RNP delivery vehicle with minimal RNA off-target activity, without the need to use the ABE mutants with reduced RNA off-target activities.8,9,13–15 ABEs do not show detectable guide-independent DNA off-target activities.10,11 This development greatly reduces the safety risks caused by ABE's guide-independent RNA off-target activities, and enables efficient and safe delivery of ABE RNPs.
We did not examine ABE's guide-dependent DNA off-targets. Others have reported that increasing the amount of Cas9 in cells increases off-target activities. 21 Our VLP-mediated ABE RNP delivery method delivers as little as 1/10 RNPs to each cell compared to current typical RNP electroporation protocols. 39 This low amount of transiently expressed ABE RNPs delivered by VLPs should also achieve reduced guide-dependent DNA off-target activities. Recently, a new ABE, ABE8e, was reported to have greatly improved on-target activity but also slightly increased DNA and RNA off-target activities. 18 Our LV capsid-based ABE RNP delivery method could be especially suitable for delivering this new version of ABE.
Conclusion
ABE RNPs show guide-dependent DNA base editing but undetectable guide-independent RNA off-target activities. ABE RNPs can be efficiently and functionally packaged into LV capsids. VLP-delivered ABE RNPs show high on-target DNA base editing activities and undetectable RNA off-target activities.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.