
Abstract
Transient expression of the CRISPR-Cas9 machinery is desirable to reduce the risks of off-targets and immune responses. Electroporation of Cas9 ribonucleoproteins (RNPs) is the most common delivery method to achieve transient Cas9 expression. Recently, retroviral capsids have been used for delivering Streptococcus pyogenes Cas9 RNPs, in which Cas9 was fused to the viral proteins. The fusion strategy may cause relative low capsid assembly efficiency. We recently developed virus-like particles (VLPs) consisting of lentiviral capsid and Staphylococcus aureus Cas9 RNPs using the specific interactions between aptamer and aptamer-binding protein (ABP), and obtained near-normal capsid assembly efficiency. Here we test whether highly active Streptococcus pyogenes Cas9 (SpCas9) RNP VLPs can be generated with high efficiency by aptamer/ABP interaction. We found that by optimizing the locations and types of aptamer used for single guide RNA modification, highly active SpCas9 RNP VLPs can be generated efficiently. VLP-delivered SpCas9 generated lower off-target insertions and deletions than SpCas9 RNPs delivered by electroporation. VLPs containing Cas9 from different species and targeting multiple genes can be efficiently prepared in single-particle preparation. Multiple-target VLPs were more efficient than the combination of single-target VLPs for simultaneous targeting of multiple genes. Thus, in addition to better safety features, the Cas9 VLPs are especially suited for multiplex genome editing. In summary, our VLPs offer safe, efficient, and flexible multiplex genome editing.
Introduction
CRISPR-Cas9 off-target rates increase with extended nuclease expression 1 and increasing Cas9 amount. 2 Reducing the nuclease action duration via delivering Streptococcus pyogenes Cas9 (SpCas9) ribonucleoproteins (RNPs) improved Cas9 specificity. 3 Currently, the most common Cas9 RNP delivery method is electroporation. 3 Typically, 10–20 μg of Cas9 protein was used for an electroporation experiment. 4
Retro- and lentiviral capsids have been used for SpCas9 RNP delivery.5–7 Two of the studies fused the SpCas9 protein with viral capsid proteins, which greatly impaired capsid assembly efficiency (three to five times lower than normal lentiviral assembly) and required typically multiple-fold concentration of the supernatants before use.5,6 The low virus-like particle (VLP) assembly efficiency makes it challenging to generate sufficient particles for various applications. SpCas9 was fused with Vpr protein and encapsulated into lentiviral capsids via Vpr/p6 interaction, the same particle also provides single guide RNA (sgRNA) through a mechanism of lentiviral vector. 7 This strategy may have normal capsid assembly efficiency although the sgRNA expressing vector may still integrate even when the integrase is mutated.
Recently, our group developed VLPs composed of lentiviral capsids and Staphylococcus aureus Cas9 (SaCas9) RNPs, and used the particles for efficient genome editing. 8 These particles packaged SaCas9 RNPs into lentiviral capsids via the interactions between aptamer and aptamer-binding protein (ABP). For RNP packaging, we used ABP Com, a 62 amino-acid-residue RNA-binding protein from bacteriophage Mu. Com specifically binds to a short RNA aptamer (named as com with core sequence “GAAUGCCUGCGAGCAUCC”) originally found in mom RNA.9,10 This ABP/aptamer pair has been used in dCas9-mediated transcription regulation. 11
We found that Com/com was the most efficient ABP/aptamer pair for packaging SaCa9 RNPs into lentiviral capsids.
8
Specifically, we fused ABP Com with lentiviral
Compared with delivering RNPs by electroporation, our lentiviral capsids typically delivered at least 10 times less Cas9 proteins than electroporation for the same number of cells.4,8 We hypothesized that the reduced RNPs used could result in improved specificity. SpCas913 is the first and most commonly used designer nuclease for genome editing.14–17 Improving SpCas9 RNP packaging efficiency by viral capsids will make the system more user-friendly. Considering the high capsid assembly and gene editing efficiency using the aptamer/ABP strategy to package SaCas9 RNPs, 8 it is attempting to explore the aptamer/ABP strategy for SpCas9 RNP package and delivery to enable more flexible editing.
Here we asked whether SpCas9 RNP VLPs can be generated with the aptamer/ABP strategy to improve particle generation efficiency, and whether the reduced SpCas9 RNPs used results in lower off-target activity than SpCas9 RNP electroporation.
Methods
Plasmids
pMD2.G (#12259; Addgene), pU6-sgRosa26–1_CBh-Cas9-T2A-BFP (#64216; Addgene), pLH-sgRNA1 (#75388; Addgene), and psPAX2-D64V (#63586; Addgene) were purchased from Addgene and have been described previously. Other plasmids were generated by this group as described in Supplementary Table S1 or as described recently.8,18 Plasmids will be made available through Addgene (IDs: 132553, 132555 and 136269) or on request. Gene synthesis was done by GenScript, Inc. All constructs generated were confirmed by Sanger sequencing. Sequence information for primers and oligos is listed in Supplementary Table S2. SpCas9 and SaCas9 target sequences and the oligos used for making the sgRNA expression constructs are listed in Supplementary Table S3.
Generation of green fluorescent protein-reporter cells
The HEK293T-derived enhanced green fluorescent protein (EGFP) reporter cells for the detection of insertions and deletions (INDELs) in HBB sickle mutant sequence and IL2RG target sequences have been described previously. 19 To generate green fluorescent protein (GFP)-reporter cells for detecting INDELs in human CLCN5 exon 2 or DMD exon 53 target sequences (see Supplementary Table S3 for target sequences), HEK293T cells were transduced with lentiviral vectors expressing the reporter cassette for human DMD exon 53 (pSin-EF2-DMD53-GFP; Supplementary Fig. S3) or CLCN5 (pSin-EF2-hCLCN5-GFP; Supplementary Fig. S4). The cells were cultured in medium with 2 μg/mL puromycin for 1 week to kill the cells negative for viral genome.
The GFP-reporter cells expressed no EGFP due to the disruption of the EGFP reading frame by the insertion of the target sequences between the start codon and the second codon of EGFP coding sequence. INDELs in targets caused by gene editing may restore the reading frame and enable GFP expression. For clarification, we call these GFP-reporter cells as HBB-IL2RG reporter, CLCN5 reporter, and DMD reporter, respectively.
Detecting gene editing activities by GFP reporter assays
The GFP reporter cells were used to detect gene editing activities of the respective SaCas9 or SpCas9 RNPs. HBB-IL2RG reporter cells were used to detect INDELs targeting human beta hemoglobin (HBB) sickle-cell mutation sequence or human IL2RG target sequence in the GFP-reporter cassette. The CLCN5 reporter cells were used to detect INDELs targeting CLCN5 exon 2. DMD reporter cells were used to detect INDELs targeting DMD exon 53 by SaCas9 or SpCas9 RNPs. GFP-positive cells were analyzed by fluorescence microscopy or flow cytometry (Accuri C6; BD Biosciences) as described previously. 18 These cells were developed for conveniently and quantitatively detecting gene editing activity to optimize our Cas9 delivery system. Gene editing activity was also essayed by next-generation sequencing (NGS) and Tracking of Indels by Decomposition (TIDE), as described in the following sections.
Lentiviral vector and Cas9 VLP production
Lentiviral vector plasmid pLH-sgRNA1-IL2RG (expressing IL2RG targeting sgRNA) was used to produce integration-defective lentiviral vector (packaged by packaging plasmid pspAX2-D64V) as described. 19 Cas9 VLPs were produced as described recently. 8 Briefly, 13 million HEK293T cells were cultured in a 15-cm dish with 15 mL Opti-MEM. Sixteen micrograms of ABP-modified packaging plasmid pspAX2-D64V-NC-ABP (ABP could be MCP [MS2 coat protein, binding to MS2], PCP [PP7 coat protein, binding to PP7], λ N22 peptide [binding to BoxB] or Com [binding to com]), 6 μg envelope plasmid (pMD2.G), and 16 μg plasmid DNA coexpressing SpCas9 and the aptamer-modified sgRNA were mixed in 1 mL Opti-MEM. Seventy-six microliters of 1 mg/mL polyethylenimine (PEI; Polysciences Inc.) was mixed in 1 mL Opti-MEM. The DNA mixture and the PEI mixture were then mixed and incubated at room temperature for 15 min. The DNA/PEI mixture was then added to the cells in Opti-MEM.
Twenty-four hours after transfection, the medium was changed into 15 mL Opti-MEM and the Cas9 VLPs were collected 48 and 72 h after transfection. The supernatant was spun for 10 min at 500 g to remove cell debris. The cleared supernatant could be used directly or be further processed as described below. Transfection can also be done in 10-cm dishes or six-well plates with FuGENE HD. DNA amount was proportionally scaled down according to vessel surface area. FuGENE HD was more efficient than PEI when transfecting multiple plasmids, but PEI was used for large-scale transfection for cost consideration.
Concentrating lentiviral vectors and Cas9 VLPs
The supernatant containing lentiviral vectors or Cas9 VLPs was concentrated with the KR2i TFF System (KrosFlo® Research 2i Tangential Flow Filtration System) (Cat. No. SYR2-U20; Spectrum Lab) using the concentration/diafiltration/concentration mode. Briefly, 150–300 mL of supernatant was first concentrated to about 50 mL, diafiltrated with 500–1000 mL of phosphate-buffered saline, and finally concentrated to about 8 mL. The hollow fiber filter modules were made from modified polyethersulfone, with a molecular-weight cutoff of 500 kDa. The flow rate and the pressure limit were 80 mL/min and 8 psi for the filter module D02-E500-05-N, and 10 mL/min and 5 psi for the filter module C02-E500-05-N. Lentiviral vectors and Cas9 VLPs were also concentrated by ultracentrifugation as described previously. 18
Lentiviral vector and Cas9 VLP quantification
Concentrations of lentiviral vectors and Cas9 VLPs were determined by p24 (a capsid antigen)-based ELISA (QuickTiter™ Lentivirus Titer Kit Catalog Number VPK-107; Cell Biolabs). When unconcentrated samples were assayed, the viral particles were precipitated according to the manufacturer's instructions so that the soluble p24 protein was not detected.
Western blotting analysis of Cas9 and capsid proteins in VLPs
Two hundred nanograms p24 of VLPs were transiently treated with 0.5% Triton X-100 following a published procedure. 20 Briefly, particles were centrifuged with a Sorvall T-890 rotor (2 h at 120,000 g) through step gradients containing a 1 mL layer of 10% sucrose in STE (100 mM NaCl, 50 mM Tris/HCl [pH 7.5], and 1 mM EDTA) with or without 0.5% Triton X-100, and a cushion of 2 mL 20% sucrose in STE solution. Pelleted particles were directly lysed in 100 μL of 1 × Laemmli sample buffer for Western blotting or for purifying RNA for quantitative reverse transcription (PCR RT-qPCR) analysis.
The proteins in each sample were separated on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed by Western blotting. The antibodies used include mouse monoclonal anti-SpCas9 antibody (CRISPR-Cas9 monoclonal antibody [7A9-3A3], Cat No. MA1-201; Thermo Fisher) and p24 mouse monoclonal antibody for capsid protein (CA; Cat No. 310810, 1:1000; Cell Biolabs). Horseradish peroxidase-conjugated anti-mouse IgG (H+L) (Cat No. 31430, 1:5000; ThermoFisher Scientific) and anti-rabbit IgG (H+L) (Cat No. 31460; 1:5000) secondary antibodies were used in Western blotting. SpCas9 RNP standards were GenCrispr NLS-Cas9-NLS nuclease from GenScript (Cat No. Z03389S). Chemiluminescent reagents (Pierce) were used to visualize the protein signals under the LAS-3000 system (Fujifilm). Densitometry (NIH ImageJ) was used to quantify protein amount.
RNA isolation and RT-qPCR analyses
An miRNeasy Mini Kit (Cat No. 217004; QIAGEN) was used to isolate RNA from concentrated particles or cells. The QuantiTect Reverse Transcription Kit (QIAGEN) was used to reverse-transcribe the RNA to cDNA. For sgRNA reverse transcription, 0.6 μL random primers provided in the kit and 0.4 μL sgRNA-specific primers (Sp-sgRNA-R1, gcaccgactcggtgccactt, 20 μM) were used for reverse transcription. Then guide-specific forward primers (Supplementary Table S2) were used together with Sp-sgRNA-R1 in SYBR green-based RT-qPCR to detect specific sgRNAs. Polymerase chain reaction (PCR) was run on a QuantStudio 3 or ABI 7500 instrument.
Lentiviral vector and VLP transduction
Concentrated lentiviral vectors or VLPs (equivalent to 10–300 ng of p24 protein) were added to 2.5 × 104 cells grown in 24-well plates, with 8 μg/mL polybrene. Unconcentrated particle-containing supernatant was diluted with fresh medium to transduce cells. The cells were incubated with the particle containing medium for 12–24 h, after which the medium was replaced with normal medium.
Examination of Cas9 protein degradation in cells
A total of 2 × 104 HEK293T cells were transduced with 100 ng p24 of IL2RG targeting VLPs with or without aptamer. Twelve hours after transduction, the cells were maintained in Opti-MEM to limit cell division. Fresh medium was changed every 48 h. Cells were collected every 12 h after transduction to detect the presence of SpCas9 protein by Western blotting. The relative expression of Cas9 was quantified by densitometry with NIH ImageJ software (Version 1.49). The densitometry data were used to determine protein half-life using the one-phase decay method of GraphPad Prism 5.0.
Cas9 RNP electroporation
CRISPR RNA for HBB sickle mutation (GrUrArArCrGrGrCrArGrA rCrUrUrCrUrCrCrArCrGrUrUrUrUrArGrArGrCrUrArUrGrCrU) was synthesized by IDT Inc., Alt-R®CRISPR-Cas9 tracrRNA, Alt-R® CRISPR-Cas9 Negative Control crRNA, Alt-R® Cas9 Electroporation Enhancer, and Nuclease Free Duplex Buffer were purchased from IDT, Inc. RNP reconstitution and electroporation were performed following the instruction of IDT (https://sfvideo.blob.core.windows.net/sitefinity/docs/default-source/protocol/alt-r-crispr-cas9-user-guide-ribonucleoprotein-electroporation-amaxa-nucleofector-system6a01611532796e2eaa53ff00001c1b3c.pdf?sfvrsn=71c43407_30).
A total of 2 × 105 HEK293T cells were used for each electroporation with the Amaxa Nucleofector system (Lonza). The cells were resuspended in 100 μL of nucleofection buffer from the Cell Line Nucleofector™ Kit V (Cat. No.: VCA-1003; Lonza), and put in the electroporation cuvette. Then 1 μL of Alt-R® Cas9 Electroporation Enhancer and 5 μL of reconstituted SpCas9 RNPs were added to the cells in the cuvette. Finally, the cells were given an electric shock with protocol Q-001. The cells were removed from the cuvette and cultured in growth medium for 48 h before analysis.
Next-generation sequencing and data analysis
The following primers were used for PCR amplifying the target regions for targeted next-generation sequencing (Amplicon-EZ; GENEWIZ, Morrisville, NC, USA): Primers HBB-R1 and HBB-R3 for the endogenous HBB target sequence, IL2RG-F2 and IL2RG-3301R for the endogenous IL2RG target sequence, DMD 53-F and DMD 53-R for the DMD exon 53 target sequence, hCLCN5-F and hCLCN5-R for the endogenous CLCN5 target region, and Reporter-F and Reporter-R1 for the target sequences in all the three GFP reporter cassettes (see Supplementary Table S2 for primer sequences). The proofreading HotStart® ReadyMix from KAPA Biosystems (Wilmington, MA) was used for PCR. Usually, 50,000 reads/amplicon were obtained. Analysis of INDELs was done with the online software Cas-Analyzer 21 and CRISPRESSO2. 22
Gene editing analysis by TIDE or Inference of CRISPR Edits
Efficiency of gene editing was also estimated by TIDE 23 (http://shinyapps.datacurators.nl/tide/) or Inference of CRISPR Edits (ICE) (https://ice.synthego.com/#/) using Sanger sequencing traces. When editing efficiency are >20%, the two methods give similar results.
Statistical analyses
GraphPad Prism software (version 5.0) was used for statistical analyses. T-tests were used to compare the averages of two groups. Analysis of variance (ANOVA) was performed followed by Tukey post hoc tests to analyze data from more than two groups. Bonferroni post hoc tests were performed following ANOVA in cases of two factors. Bivariate fit analyses were done with JMP software. p < 0.05 was regarded as statistically significant.
Results
Aptamer addition in sgRNA enables formation of functional SpCas9 RNP containing VLPs via aptamer and ABP interaction
Since using the fusion strategy to package SpCas9 RNPs into viral capsids showed low capsid assembly efficiency,5,6 and using the aptamer/ABP strategy to package SaCas9 RNPs showed near-normal capsid assembly efficiency, 8 we asked whether SpCas9 RNPs can be packaged into lentiviral capsids via aptamer/ABP interaction. SpCas9 and SaCas9 have different trans-activating crRNAs scaffolds, and previous attempts of including aptamer into SpCas9 sgRNA greatly decreased the nuclease activity of SpCas9 RNPs.24,25 Although we recently observed that replacing the tetraloop of sgRNA with com aptamer was able to package and deliver SpCas9 RNPs, 26 we decided to systematically screen for the types of aptamer and positions of insertion that best preserve the activities of SpCas9 RNPs.
We tested four different types of aptamers, including MS2, 27 PP7, 28 BoxB, 29 and com 10 (Fig. 1A). Each aptamer was tested at three different locations: (1) replacing the tetraloop; (2) replacing the loop of stem loop 2 (ST2); and (3) inserting at the 3′ end of the sgRNA. The minimal aptamer sequences used and the positions tested were based on previous works of modifying sgRNAs for CRISPR-based gene regulation and DNA labeling.11,30,31 We only tested one copy of aptamer since it was found that inserting more than one copy of aptamer in sgRNA greatly decreased the expression 11 and that sgRNA is a limiting factor for Cas9 activity.32,33
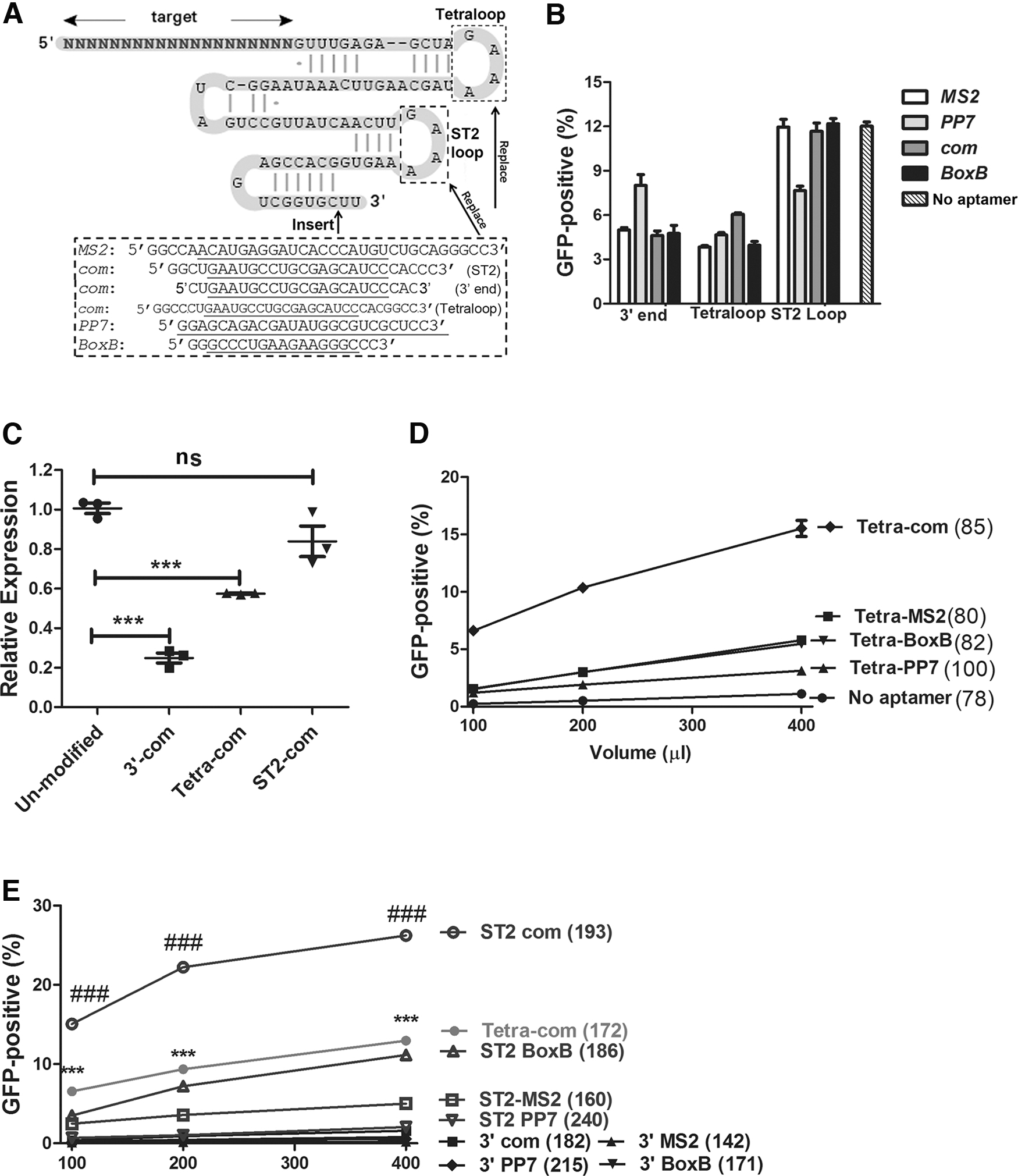
Screening for aptamer and location for most efficient packaging of SpCas9 RNPs by lentiviral capsids.
To have a sensitive assay to quantitatively compare gene editing activities, we used the GFP-reporter cells we described previously. 19 These HEK293T-derived GFP-reporter cells were integrated with a GFP-reporter cassette, where a stretch of 119 bp HBB/IL2RG target sequences were inserted after the GFP start codon ATG. The cells did not express EGFP due to the insertion disrupting EGFP reading frame. Gene editing resultant INDELs in HBB/IL2RG target sequence may restore the EGFP reading frame, resulting in EGFP expression.
When plasmid DNA expressing SpCas9 and various modified IL2RG-targeting sgRNAs were transfected into the HBB/IL2RG GFP-reporter cells, we found that the aptamer at the 3′ end or the tetraloop impaired sgRNA activity, whereas MS2, com, and BoxB aptamer at the ST2 loop did not impair sgRNA activity (Fig. 1B). We compared the static levels of com-modified sgRNAs in HEK293T cells, and observed that the gene editing activities were correlated to sgRNA levels: high sgRNA expression corresponded to high gene editing activities (Fig. 1C). Note that although the amplicons were slightly different due to variant aptamer and position, they had very similar standard curves (Supplementary Fig. S1).
We then tested the best aptamer and position combination for most efficient generation of SpCas9 RNP VLPs. We packaged SpCas9 RNPs into lentiviral capsids by cotransfecting the following three plasmids into HEK293T cells: the envelope plasmid pMD2.G, the target plasmid coexpressing SpCas9 and various aptamer-modified IL2RG-targeting sgRNAs, and the packaging plasmids modified by the corresponding ABPs (pspAX2-D64V-NC-ABP) as described recently.8,18 The ABP-modified packaging plasmids have MCP (binding to MS2), 27 PCP (binding to PP7), 28 λ N22 peptide (binding to BoxB), 29 or Com (binding to com) 10 inserted after the second zinc finger domain of NC protein. The supernatants containing various capsid-RNPs were used to transduce HBB-IL2RG GFP-reporter cells to test their genome editing activities.
Flow cytometry analyses found that replacing the ST2 loop with com aptamer generated the most GFP-positive cells (Fig. 1D, E), replacing the tetraloop with com or BoxB aptamer followed. The rest of the combinations generated very few GFP-positive cells. Note that although the sgRNA without aptamer modification was very active in transfection experiments (Fig. 1B), it had hardly any activity in packaging and transduction experiments (Fig. 1D). This lack of activity suggested that the observed gene editing activity from aptamer-modified sgRNAs could not be attributed to residual plasmid DNA. Although replacing the ST2 loop of sgRNA with MS2, com, and BoxB all preserved sgRNA activity in transfection experiments, aptamer com and ABP Com combination enabled the most efficient generation of active SpCas9 RNP VLPs.
NGS verified that the GFP expression observed in these assays was the result of genome editing. Seventy-five nanograms p24 of RNPST2-com VLPs generated 65.5% INDELs in the IL2RG target sequence, whereas similar amounts of RNPTetra-com VLPs generated 23.7% INDELs in the target sequence (Supplementary Fig. S2), confirming better activity when aptamer was at the ST2-loop. Thus, these experiments found the best aptamer position (ST2 loop) and the best aptamer/ABP pair (com/Com) for most efficient packaging of SpCas9 RNPs into lentiviral capsids.
One of the major purposes for using the aptamer/ABP strategy to package SpCas9 into lentiviral capsids was to avoid low capsid yields observed with the fusion strategy. 5 We compared the particle assembly yields of SpCas9 RNPs with that of SaCa9 RNPs, and found that they were 116.2 ± 1.2 ng/mL p24 for SaCas9 and 132.8 ± 2.0 ng/mL p24 for SpCas9 (N = 3, t = 6.880, df = 4, p = 0.0023, two-tailed t-test). Compared with normal lentivirus (LV) production yields (set as 100%), capsid assembly efficiency for SaCa9 RNPs and SpCas9 RNPs was 88.9% ± 1.0% (n = 6) and 101.5% ± 1.5% (n = 3), respectively. Thus, the capsid assembly efficiency of SpCas9 RNPs was similar to that of normal LV. This is in sharp contrast to a three to five times of capsid assembly efficiency reduction when packaging SpCas9 RNPs using the fusion strategy. 5
Functional RNP packaging into capsids depended on aptamer/ABP interaction
To examine whether aptamer/ABP interaction was necessary for functional packaging of SpCas9 RNPs, we compared the SpCas9 protein content in capsids prepared with IL2RG-targeting sgRNA, with and without aptamer modification. Since the capsid-RNPs were concentrated with the tangential flow filtration (TFF) system, extracellular vesicles and large protein complexes could also be retained. To eliminate possible SpCas9 protein associated with membranous structures, we transiently treated the particles with 0.5% Triton X-100 buffer, 20 a procedure shown to successfully remove membrane-associated SaCas9 protein in our previous study. 8 We confirmed that this procedure greatly reduced capsid protein (p24; Fig. 2A). However, it did not reduce SpCas9 protein in RNP preparations with or without com aptamer. The data suggested that SpCas9 in the preparations was resistant to transient 0.5% Triton X-100 treatment.

Aptamer/ABP interaction is necessary for functional SpCas9 delivery.
We then compared the amount of SpCas9 protein in preparations with and without aptamer modification of sgRNA. Contradictory to lack of gene editing activities in preparations with unmodified sgRNA (Fig. 1D), we observed even more SpCas9 protein in preparations with unmodified sgRNA (no com) than in preparations with com modification (Fig. 2A). In both preparations, the full-length Cas9 protein was the most abundant, although weaker and smaller bands, possibly from partially degraded Cas9, were also visible. Compared with SpCas9 of known concentration, we estimated that 1 ng p24 of capsids contained 3 ng of SpCas9 RNPs.
We did RT-qPCR to check whether there was sgRNA in the VLP preparations, and detected 678.1 ± 216.4 (N = 3, without com) and 205.5 ± 55.40 (N = 3, with com) copies/capsid, respectively. Although no statistical difference was observed due to large variation (p = 0.1018), the average sgRNA copies/capsid were even larger in VLP preparation with unmodified sgRNA than in that with com-modified sgRNA. Apparently, sgRNA deficiency could not explain the lack of gene editing activity in aptamer-negative RNPs.
In agreement with this observation, providing extra IL2RG sgRNA by cotransducing an IL2RG sgRNA-expressing lentiviral vector with the VLPs only slightly increased the gene editing activity of aptamer-negative RNPs, but did not increase the gene editing activity of aptamer-positive RNPs (Fig. 2B). The IL2RG sgRNA-expressing lentiviral vector was functional since it generated >10% GFP-positive cells when transducing GFP reporter cells transfected with SpCas9 expressing DNA. Even after supplementing with sgRNA expressed from lentiviral vectors, the gene editing activity of the aptamer-negative RNPs was still less than half of that of aptamer-positive RNPs. These data suggested that sgRNA deficiency was not the main reason for lack of gene editing activity from aptamer-negative RNPs.
To further examine the discrepancy, we transduced VLPs containing aptamer-positive and aptamer-negative IL2RG RNPs into HEK293T cells and checked the Cas9 protein level every 12 h. Although the same amounts of capsids (100 ng p24) were transduced, the level of full-length Cas9 protein from aptamer-negative RNPs was less than 20% of that from aptamer-positive RNPs 12 h after transduction (Fig. 2C, D). In addition, strong signals of smaller bands were observed in cells treated with aptamer-negative RNPs. These signals were much weaker in cells treated with aptamer-positive RNPs, and not observed in untransduced cells, suggesting that they were signals of partially degraded Cas9 protein.
The data revealed two SpCas9 populations in our VLP preparations: a nonfunctional and a functional RNP population. The nonfunctional RNPs were resistant to transient Triton X-100 treatment but were quickly degraded after applying to the cells (mainly in the aptamer-negative RNPs). The nature of the nonfunctional Cas9 RNPs in the preparations was unclear. Recently, Montagna et al. also observed abundant nonfunctional SpCas9 protein in their extracellular vesicle (EV) preparations after Cas9 overexpression. 34 Our TFF concentration method could not separate lentiviral particles from EVs. Thus the nonfunctional SpCas9 proteins in the two studies could have similar identity. The functional SpCas9 RNPs were aptamer dependent and protected from rapid degradation. These RNPs most likely explained the observed gene editing activity (mainly in the aptamer-positive RNPs). They were most likely packaged into the capsids through aptamer/ABP interaction, and thus have a relatively longer half-life (∼7.3 h) than the nonfunctional RNPs with half-life of about 3 h. The 7.3-h half-life conferred gene editing activity but also ensured transient nuclease expression.
The data showed that although the presence of SpCas9 protein or RNPs in the preparations was not aptamer/ABP dependent, the functional packaging and delivery of RNPs were.
Efficient genome editing by SpCas9 VLPs
We examined the gene editing activities of the RNP-loaded VLPs on various target genes in HEK293T-derived reporter cells. In addition to HBB-IL2RG GFP-reporter cells, 19 we also used DMD GFP reporter cells (Supplementary Fig. S3) 26 and CLCN5 GFP reporter cells (Supplementary Fig. S4). These reporter cells were generated for sensitively detecting INDELs in their respective target sequences by flow cytometry. It also enabled us to compare the gene editing efficiencies on lentiviral-integrated targets and endogenous targets.
Delivering 100 ng of CLCN5-targeting RNPs per 104 CLCN5-GFP reporter cells generated 85.4% INDELs on the lentiviral-integrated CLCN5 target sequence (Fig. 3A), and 37.4% INDELs on the endogenous CLCN5 gene in these cells (Supplementary Fig. S5A). Delivering 100 ng of IL2RG-targeting RNPs per 104 cells generated 77.1% INDELs on the lentiviral-integrated IL2RG target site (Fig. 3B) and 8.1% on the endogenous IL2RG gene of the same cells (Supplementary Fig. S5B). Delivering 120 ng of DMD-targeting RNPs per 104 cells generated 59.2% INDELs on the lentiviral-integrated DMD target site (Supplementary Fig. S5C), whereas a double amount of RNPs to the same number of HEK293T cells generated 25.0% INDELs on the endogenous DMD target (Supplementary Fig. S5D). Thus, for these endogenous loci that may not be expressed in HEK293T cells, lower gene editing efficiencies were observed than for the lentiviral-integrated targets. More Cas9 protein would be needed to obtain higher gene editing efficiency on these endogenous loci.
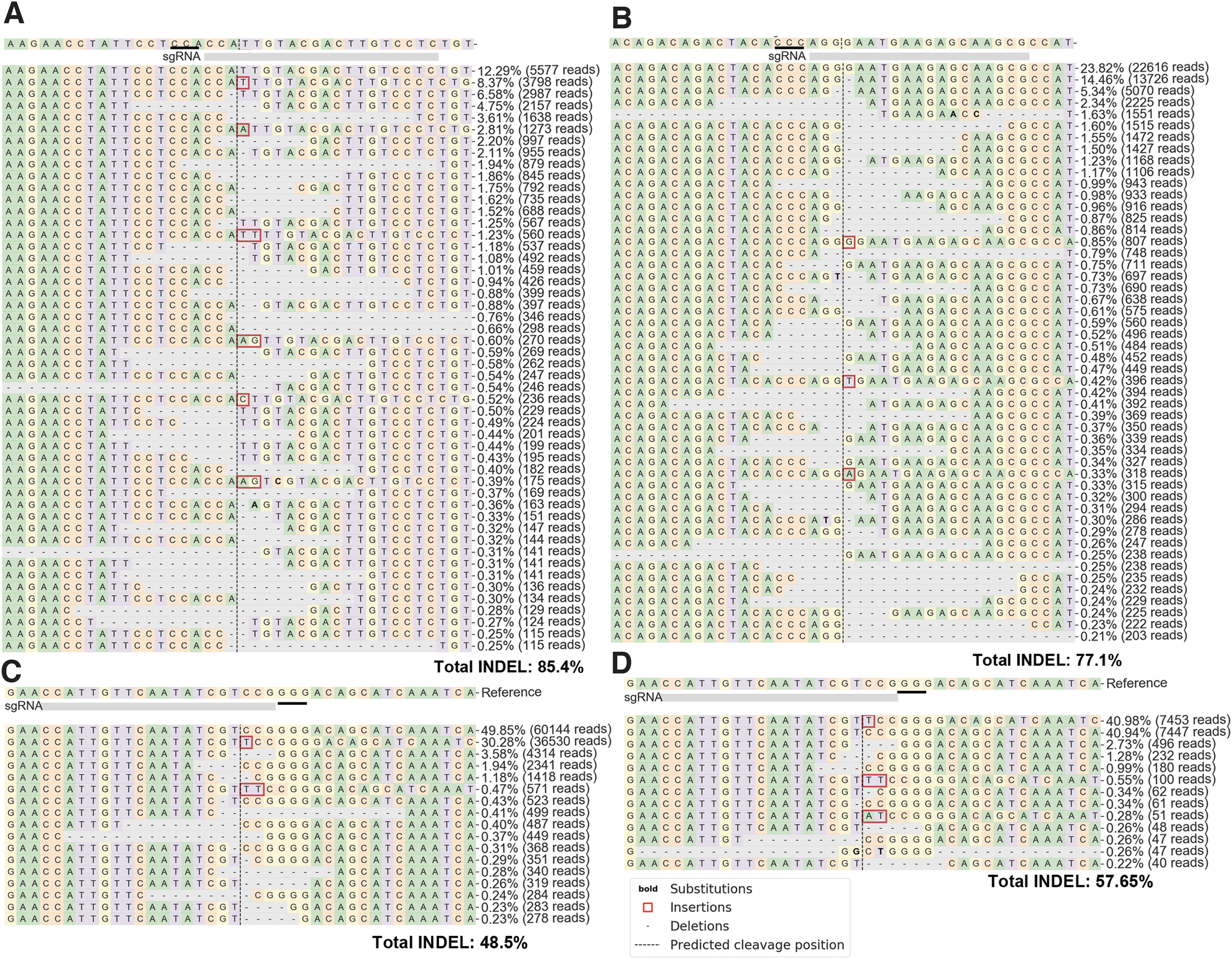
Gene editing activities of VLP-delivered SpCas9 RNPs.
We then targeted actively expressed genes such as TP53 and GAPDH in HEK293T cells. We observed 48.5% INDELs in TP53 (Fig. 3C) and 38.6% INDELs in GAPDH (Supplementary Fig. S6A) at a dose of 80 ng of RNPs per 104 cells. The efficiencies of targeting these actively expressed genes were apparently higher than those targeting the inactive genes examined above. We also targeted an intergenic region on chromosome 20 (GRCh38.p13, 32752960 to 32752979) with 80 ng of RNPs per 104 cells and observed 57.5% INDELs (Supplementary Fig. S6B).
We then used these VLP-delivered RNPs to target the endogenous sites in MDA-MB-231 cells that are known hard to transfect. At a dose of 160 ng of RNPs per 104 cells, we observed 57.65% INDELs in TP53 (Fig. 3D) and 44.7% INDELs in GAPDH (Supplementary Fig. S6C). Thus, the VLPs can efficiently deliver RNPs to different human cells and target different loci. In all of these experiments, unconcentrated VLP-containing supernatants were used.
Cas9 VLPs showed higher on-target activity and lower off-target activity than electroporation-delivered Cas9 RNPs
Based on Western blotting data, the highest Cas9 RNPs delivered by VLPs were 24 pg/cell (200 ng p24 of capsids, equivalent to 600 ng of RNPs, for 2.5 × 104 human cells). This dose was lower than half of the Cas9 protein used in common electroporation protocols (50–100 pg/cell). We wondered whether the less Cas9 protein delivered by VLPs resulted in lower off-target rates. To compare the off-target activities between Cas9 VLPs and electroporation-delivered Cas9 RNPs, we again used our HBB/IL2RG GFP-reporter cells. 19 These cells were integrated with GFP-reporter cassette containing the sickle cell disease mutation that is one nucleotide different than the endogenous HBB sequence (Fig. 4A). The single-nucleotide mismatch between the sgRNA (targeting the sickle mutant sequence) and the endogenous HBB sequence allows for a low efficiency of “off-target” editing that can be detected and compared between different delivery methods.
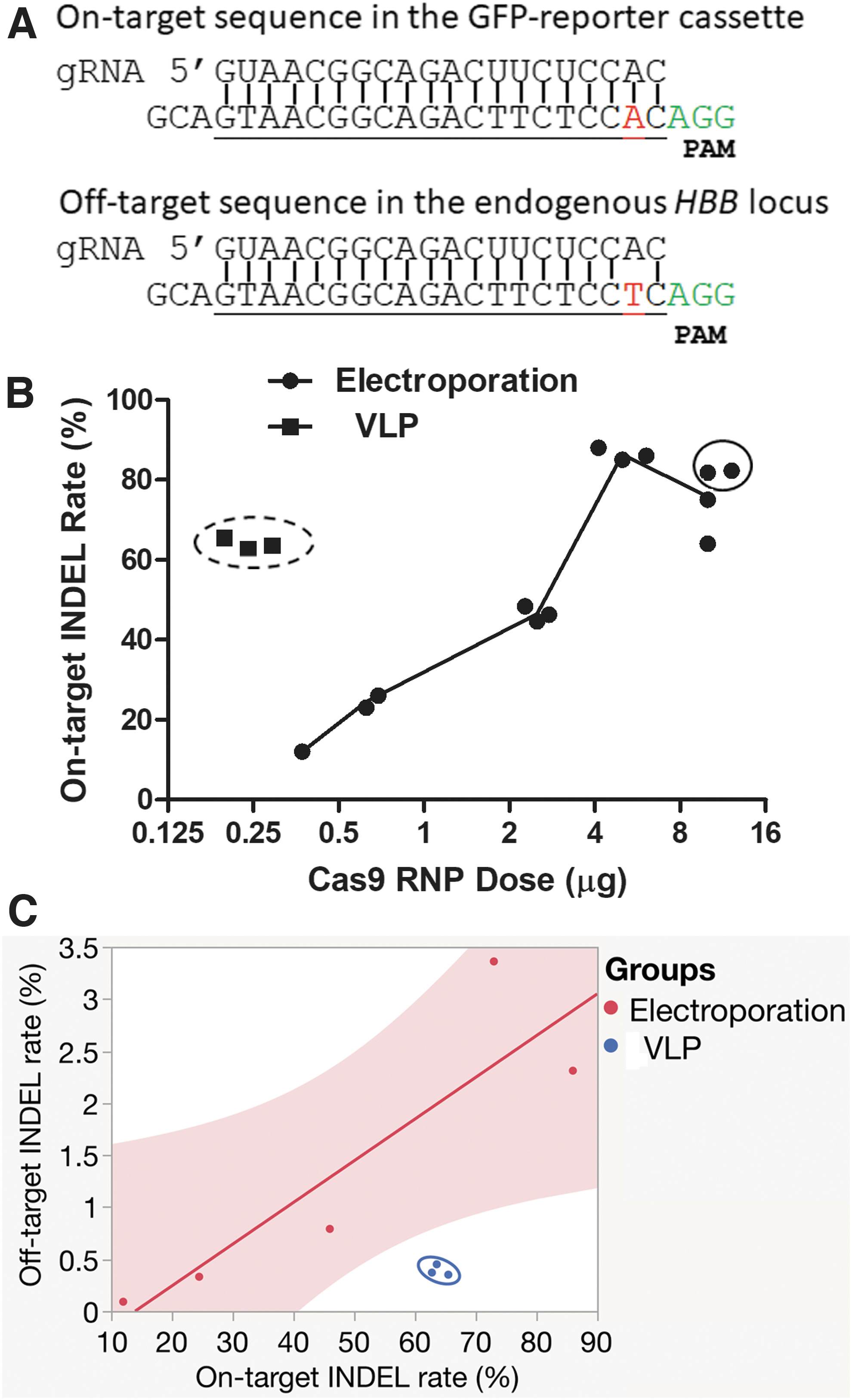
On-target and off-target comparison between Cas9 RNPs delivered by VLPs and electroporation.
We compared VLP- and electroporation-delivered SpCas9 RNPs targeting the sickle cell disease mutation. In this setting, INDELs in the GFP-reporter cassette reflected on-target gene editing activity due to the 100% match between the target sequence and the sgRNA, whereas INDELs in the corresponding endogenous HBB gene reflected off-target activity due to the one nt mismatch with the sgRNA.
We delivered the RNPs to the cells by VLPs or by electroporation. After electroporation of variant amounts of sickle mutation targeting RNPs, we observed increasing on-target INDELs with increasing amount of recombinant RNPs until it reached a plateau at 5 μg (Fig. 4B). We transduced various amounts of sickle mutant-targeting VLPs into the reporter cells and found that 80 ng p24 of Cas9 VLPs (∼0.24 μg of total Cas9) produced a similar percentage of GFP-positive cells as electroporation of 5 μg of RNPs. We thus determined the on-target INDELs by TIDE and performed NGS analysis on part of the samples for confirmation.
We found that less than 1/10 Cas9 RNPs delivered by VLPs (Fig. 4B, dots in dashed circle) generated similar on-target INDEL rates of RNPs delivered by electroporation. Bivariate fit of on-target INDEL rate by RNP dosage showed that VLP-delivered RNPs significantly deviated from RNPs delivered by electroporation (p = 0.022; Supplementary Fig. S7). Apparently, VLP delivery showed better unit activity than electroporation.
We then examined INDEL rates by NGS in the endogenous HBB target to compare off-target INDELs. RNPs delivered by electroporation generated increasing off-target INDELs with increasing on-target INDEL rates (Fig. 4C) and RNP dosage, consistent with previous finding that the amount of Cas9 protein affects off-target activity. 2 However, off-target INDELs from VLP-delivered RNPs clearly deviated from those of electroporation-delivered RNPs (Fig. 4C, dots in circle, p = 0.0414).
VLPs enable multiplex gene editing with single-particle preparation
Since each capsid has about 5000 copies of capsid precursor protein Gag, theoretically each capsid can package multiple Cas9 RNPs. If two genes need to be edited, the chances will be high that RNPs targeting both loci be packaged into the same capsid, as illustrated in Figure 5A. It is expected that copackaged RNPs targeting different loci would be more efficient than individually packaged RNPs for multiplex gene editing.

Copackaging of RNPs for efficient multiplex gene targeting and long-range genomic deletion.
We first tested whether VLPs loaded with SpCas9 RNPs targeting two different genes, DMD exon 53 and CLCN5 exon 2, could be generated in a single transfection experiment. We generated the two-target VLPs in the same way as generating single-target VLPs, except that the two-target plasmids with different sgRNAs were mixed 1:1 during transfection. For comparison, single-target VLPs for each gene were prepared in parallel. Eighty nanograms p24 of two-target VLPs or 40 ng each of the single-target particles was transduced into DMD and CLCN5 reporter cells for comparison. The two-target VLPs showed similar gene editing activity on the CLCN5 gene (Table 1), but a slight decrease of gene editing activity on DMD, possibly a result of competition between the two types of RNPs during particle generation. The data showed that generating two-target VLPs in single-particle preparation is possible.
Sp-CLCN5-g1 RNPs were assayed in CLCN5 GFP reporter cells, Sp-DMD53-g2 and Sa-DMD53-g2 RNPs were assayed in DMD GFP reporter cells. Due to different INDEL profiles of different sgRNAs, the activities of different sgRNAs were not comparable by this assay.
Indicates p < 0.001.
Two of the four guide RNAs (targeting DMD introns 50 and 51) were not listed in the table since there is no GFP-reporter assay for them.
Co, copackaged; GFP, green fluorescent protein; In, individually packaged; INDEL, insertions and deletions; RNP, ribonucleoprotein; SaCas9, Staphylococcus aureus Cas9; SpCas9, Streptococcus pyogenes Cas9.
We then asked whether VLPs loaded with SaCas9 and SpCas9 RNPs could be prepared in single-particle preparation. We tested generating VLPs loaded with SaCas9 RNPs targeting DMD exon 53 and SpCas9 RNPs targeting CLCN5 exon 2, similarly by mixing the two-target plasmids at a ratio of 1:1 during transfection. After applying the particles to the DMD and CLCN5 GFP-reporter cells, flow cytometry showed that for both target genes, 80 ng p24 of two-target VLPs showed similar activities as combining 40 ng p24 of each one-target VLP (Table 1). The data showed that particles containing both SaCas9 RNPs and SpCas9 RNPs could be generated in single preparation.
We further tested generating particles targeting four loci in a single particle generation: DMD intron 50 (SaCas9), DMD intron 51 (SaCas9), DMD exon 53 (SpCas9), and CLCN5 exon 2 (SpCas9). These included two SaCas9 RNPs and two SpCas9 RNPs. For generating particles targeting four genes, we mixed the four-target plasmids at a ratio of 1:1:1:1 during particle generation. We compared the activities of 160 ng p24 of four-target particles with the combination of 40 ng p24 of each one-target particle. Flow cytometry observed that compared with the using of four one-target particles, the four-target particles showed similar activities on CLCN5, and 84% activities on DMD exon 53 (Table 1). We performed NGS to analyze the INDELs in CLCN5 target sequence generated by four-target particles, and indeed observed INDELs in 64.5% of alleles (Supplementary Fig. S8), confirming gene editing. Note that this activity was obtained from unconcentrated VLP-containing culture supernatants.
The rest of the two edited loci, in DMD intron 50 and intron 51, respectively, were 2361 bp apart (Fig. 5B). We used two primers (50-F and 51-R2, 2645 bp apart) flanking the two-target sites to amplify the region between, and detected a fragment of 284 bp, but not the full-length 2645 bp (Fig. 5C), indicating complete deletion of the DNA between the two-target sites. DNA deletion was confirmed by DNA sequencing (Fig. 5D). The data showed that the four-target Cas9 VLPs were functional on all four targets.
We then examined whether generating multiple-target VLPs compromised particle generation efficiency. The capsid assembly efficiency for multiple-target VLPs was 87% of that for single-target VLPs, but the difference was not statistically significant (179.0 ± 6.8 ng/mL p24, n = 11 for single-target particles, 156.3 ± 3.4 ng/mL p24, n = 4 for multiple-target particles, p = 0.1027, Mann/Whitney test). These data showed that generating multiple-target VLPs containing SaCas9 and SpCas9 RNPs was possible.
Multiple-target VLPs are more efficient than single-target VLPs for multiplex gene editing
In multiplex gene editing, it is important that all target loci in the same cell are targeted. To test whether multiple-gene-targeting VLPs benefit cotargeting, we quantitatively compared the cotargeting of DMD intron 50 (gRNA Sa-50-g2) and DMD intron 51 (gRNA Sa-51-g2) by two-target VLPs and single-target VLPs. Simultaneous targeting both sites in the same cell will delete the DNA between the two-target sites and produce a short PCR product of 284 bp by flanking primers. To avoid DNA deletion in 100% cells (as in Fig. 5C), which prevented us from comparing efficiency, we titrated the amounts of VLPs used. We found that 10 ng p24 of two-target VLPs generated more deletions than combining 20 ng p24 of each single-target VLP (a total of 40 ng of p24, DNA band in red dashed box in Fig. 5E). The data clearly showed that multiple-target VLPs were more efficient in simultaneously targeting multiple sites in the same cell.
CRISPR-Cas9 has been shown to generate largely unpredictable large deletions.35–37 However, in chromosome engineering, defined large genomic deletions are desired. 38 In the four-target particle experiment described above, the target sites for DMD 53-sp-g2 and CLCN5 Sp-g1 were over 18 Mb away on X-chromosome (Fig. 5F). We wondered whether deletions of this size (18 Mb) could be generated. PCR was performed with primers flanking the two loci (53-R and hCLCN5-R) to detect the possible 18 Mb deletions. In cells treated with copackaged and combined individually packaged VLPs, we observed PCR products expected from alleles with deletions (Fig. 5G). The undeleted alleles could not be amplified due to their large sizes. DNA sequencing confirmed that the observed PCR products were indeed the results of the expected deletion (Fig. 5H). Note that we sequenced the PCR products from mixed alleles without cloning, observing dominant peaks after the predicted cleavage sites suggested that the cleavage sites were quite conserved in different alleles and cells. This was in sharp contrast to unpredictable large deletions caused by sgRNA action.35–37
Discussion
Here we describe a strategy to generate VLPs loaded with SpCas9 RNPs for safe and efficient genome editing. Our SpCas9 RNP-laden VLPs are superior to the current Cas9 RNP delivery methods in many aspects.
First, we used the specific aptamer/ABP interaction to package SpCas9 RNPs into lentiviral capsids to generate VLPs loaded with Cas9 RNPs. Several groups generated SpCas9 VLPs via fusing SpCas9 to capsid proteins.5,6 However, the fusion strategy greatly decreased capsid assemble efficiency to 20–30% of normal lentiviral assembly efficiency, making particle generation inefficient. We used the aptamer/ABP interaction strategy to generate SpCas9 VLPs and achieved capsid assembly efficiency of that of normal lentiviral capsid assembly. Although the same strategy was used to generate SaCaS9 VLPs in our recent study, 8 we found that the best condition for SpCas9 VLPs was different from that of SaCas9 and was only found after systematic searching for types of aptamer and locations for insertion. We were unsuccessful in generating Cas12a VLPs by the same strategy (our unpublished data), demonstrating that Cas proteins from different species have their unique characteristics.
Second, the VLP-delivered Cas9 is relatively more active and generated less off-target INDELs than electroporation-delivered Cas9 RNPs. Specifically, 1/10 Cas9 proteins delivered by VLPs were able to generate similar on-target INDEL rates as electroporation-delivered Cas9 RNPs. In addition, at similar on-target INDEL rates, the off-target INDEL rates generated by VLP-delivered RNPs were about half of that of Cas9 RNP electroporation. We reason that at least two mechanisms may underlie these observations: (1) the VLP-delivered RNPs are products of mammalian cells and do not need protein refolding necessary for biochemically purified Cas9 protein; and (2) VLP transduction may help to control the amount of Cas9 protein each cell receives, which is difficult to control in electroporation. We observed that electroporation of 10 μg of Cas9 RNPs did not increase the on-target INDEL rate but increased the off-target rate, suggesting that titrating the amount of Cas9 RNPs in electroporation may help to reduce off-targets.
Third, our method is ideal for multiplex genome editing. Three observations suggest that Cas9 RNP-laden VLPs are good delivery tools for this purpose: (1) Cas9 VLPs targeting multiple loci can be efficiently generated in single-VLP preparation; (2) VLPs containing both SaCas9 and SpCas9 RNPs can be generated in single-VLP preparation; (3) multiple-target VLPs are more efficient than combination of single-target VLPs in multiplex genome editing. Multiplex genome editing is needed in many situations, including reducing the risk of immunorejection,39–41 eradicating HIV proviral DNA, 42 and boosting cancer therapy responses. 43 Thus, the VLP system will make multiplex genome editing more feasible and efficient. The ability to use both SaCas9 and SpCas9 in the same experiment greatly increases flexibility.
It has been shown that nucleosome and heterochromatin inhibit Cas9 function,44–46 which explains why Cas9 has wide different activities against different loci. Since lentiviral-integrated targets are usually highly accessible by Cas9, 47 we developed lentiviral vector-integrated GFP-reporter cells to eliminate the efforts of target sequence accessibility on Cas9 activity for us to optimize the VLP system and to test multiplex genome editing. The observations of higher INDEL rates on lentiviral-integrated targets and lower INDEL rates on endogenous genes are consistent with these reported observations.44–47 Thus, our VLPs did not address the issue of chromatin accessibility, an issue also faced by other Cas9 delivery methods. It is shown that in situations of transient Cas9 expression and low Cas9 dosage, gene editing is more sensitive to chromatin accessibility, 48 this explains our observations of quite different editing efficiencies on the same target sequence in different chromatin contexts.
Target sequence accessibility affects Cas9 activity regardless of the delivery method. At present, there is no evidence showing that our Cas9 VLPs are different from Cas9 RNPs delivered by other methods in gene editing in the same chromatin context. Although increasing the action duration and protein amount may overcome the impeding effects of closed chromatin on gene editing to some degree, these strategies will increase safety risks and are difficult to achieve especially in in vivo applications. Thus, further work is needed to address issues related to editing of loci in heterochromatin.
In summary, the present work optimizes the conditions for using ABP/aptamer interaction to package and deliver SpCas9 RNPs by lentiviral capsids. Capsid-mediated RNP delivery showed lower off-target INDELs than RNP electroporation. SaCas9 and SpCas9 RNPs can be used in the same VLP preparation for flexible and efficient multiplex genome editing.
Data Availability
The NGS data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA590474. All data that support the findings of this study are available in the article and its Supplementary Data.
Footnotes
Author Disclosure Statement
The Regents of the Wake Forest School of Medicine has a patent pending for CRISPR-Cas9 delivery technologies on which B.L. and A.A. are inventors.
Funding Information
This work was partially supported by DOD (W81XWH2010265 to B.L.), The Bruce D. and Susan J. Meyer Charitable Fund (to B.L.) and the state of North Carolina (Grant 330054 to A.A.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.